ZYMOGRAPHY OF EXTRACELLULAR PROTEASES IN Bacillus subtilis
on
ARTICLE
ZYMOGRAPHY OF EXTRACELLULAR PROTEASES
IN Bacillus subtilis
Takeko Kodama1, Keiji Endo1, Katsutoshi Ara1, Katsuya Ozaki1 and Junichi Sekiguchi2* 1Biological Science Laboratories, Kao Corporation, 2606 Akabane, Ichikai, Haga, Tochigi 321-3497, Japan, 2Department of Bioscience and Textile Technology, Interdisciplinary Graduate School of Science and Technology, Shinshu University, 3-15-1 Tokida, Ueda, Nagano 386-8567, Japan
2Department of Bioscience and Textile Technology, Interdisciplinary Graduate School of Science and Technology, Shinshu University, 3-15-1 Tokida, Ueda, Nagano 386-8567, Japan Tel: +81-(0)268- 21-5344; Fax: +81-(0)268- 21-5345; Email:jsekigu@shinshu-u.ac.jp *Corresponding author
ABSTRACT
In Bacillus subtilis, AprE, Bpr, Epr, Mpr, NprB, NprE, Vpr and WprA have been identified as extracellular proteases. We determined protease activities from the culture supernatant of Bacillus subtilis using a 1D zymogram gel containing gelatin. The highest protease activities were found at the late stationary phase of a 75-h culture. To distinguish the proteases, the zymogram pattern of the wild type was compared with the protease-deficient mutants (aprE, bpr, epr, mpr, nprB, nprE, vpr and wprA). Activities of AprE, Bpr, Mpr, and Vpr were estimated by gelatin zymography. In addition, for the aprE mutant, the zymogram profile of Bpr was very different from that of the wild type. This suggested that AprE is involved in processing the Bpr protein.
Keywords: protease, zymography, gelatin, Bacillus subtilis
INTRODUCTION
Bacillus subtilis produces numerous extra-and intracellular proteolytic enzymes such as alkaline proteases and neutral metalloproteases (Kunst et al., 1997; Tjalsma et al., 2004). Eight extracellular proteases have been identified in B. subtilis to date, and are encoded by the following genes: aprE (Stahl and Ferrari, 1984; Wong et al., 1984), bpr (Sloma et al., 1990, Wu et al., 1990), epr (Bruckner et al., 1990; Sloma et al., 1988), mpr (Rufo et al., 1990; Sloma et al., 1990), nprB (Yang et al., 1984), nprE (Tran et al., 1991), vpr (Sloma et al., 1991), and wprA (Margot and Karamata, 1996). AprE (Alkaline (subtilisin)) and NprE (neutral protease) are the major extracellular proteolytic enzymes (Kawamura and Doi, 1984). These two enzymes account for more than 90% of the total extracellular protease activity (Kawamura and Doi, 1984). A large percentage of the remaining protease activity comes from the four serine proteases of Bpr (Bacillopeptidase F), Epr, Vpr and WprA (cell-wall-associated protease), and the two metalloproteases of Mpr and NprB.
Zymography has been used to detect proteolytic enzymes after electrophoretic
separation in gels. Active enzymes in zymogram gels are visualized as clear bands because the catalytically active proteins have proteolytically degraded the substrate in the gel. Zymography is one of the most important tools for protein identification at the nanogram scale. It has been reported that the activity of Vpr of B. subtilis was detected by fibrin zymography (Kho et al., 2005; Park et al., 2002). We also detected protease activity of AprX using protease zymography in the culture medium at the late stationary growth phase because AprX leaks outside the cell upon lysis (Kodama et al., 2007).
In this paper, we describe the determination of the activities of proteases from Bacillus subtilis with gelatin zymography. Using this method we easily detected the gelatin-degrading enzymes AprE, Bpr, Mpr and Vpr. Furthermore, the existence of multiple processed forms was observed for Bpr and Vpr. In addition, we found that the panel of Bpr activities was greatly affected by the aprE mutation. These data suggest that AprE is involved in the processing of the Bpr protein.
MATERIALS AND METHODS
Bacterial strains, plasmids and culture conditions
The B. subtilis and Escherichia coli strains used in this study are listed in Table 1. B. subtilis strains were grown in Luria-Bertani (LB) medium (1% Tryptone, 0.5% yeast extract, and 1% NaCl) or modified 2xL broth (2xL broth supplemented with 7.5% maltose and 7.5 μg/ml MnSO4). Kanamycin (10 μg/ml) was used for selection of transformants of B. subtilis strains. The B. subtilis strains were cultured in LB medium at 30ºC for 15 h, and 600 μl of the culture broth was added to 30 ml of modified 2xL broth in a Sakaguchi flask. Cells were cultured at 30ºC up to 75 h. Plasmids of pDG-Vpr, pDG-AprE, pDG-Bpr and pDG-Mpr were used for the expression of Vpr, AprE, Bpr and Mpr, respectively. E. coli strains were used as the hosts for plasmid construction and were grown in LB medium supplemented with 50 μg/ml ampicillin.
Construction of plasmids
The regions of vpr, aprE, bpr and mpr containing the Shine-Dalgarno sequences were amplified by PCR with four sets of two primers (vpr-18F: GCATGTCGACTACAAAGGGGGAAACACA and vpr+2422R: AGCTGCA TGCCTTATTC AACAGTGAAAGG, aprE-18F: GC
ATGTCGACAAAAGGAGAGG GTA
AAGA and aprE+1146R: AGCTG CAT GCTTATTGTGCAGCTGCTTG, bpr-24F: GC ATGTCGACGCATGTTGAAAAAGGGGG and bpr+4306R2: GCATGTCGACC CA CTTAATTTTCTGTGTTC, mpr-26F: GCA TGTCGACTGACACTAAAGGAGGAG and mpr+942R: AGCTGCATGCTTATTGAT TTGCCCAATA). The PCR fragments of vpr, aprE and mpr were digested with SalI and SphI, and then ligated to the corresponding sites of pDG148 (16) to generate pDG-Vpr, pDG-AprE and pDG-Mpr. The PCR fragments of bpr were digested with SalI, and then ligated to the corresponding sites of pDG148 (16) to generate pDG-Bpr. After confirming the nucleotide sequences of the cloned DNA region in pDG-Vpr, pDG-AprE, pDG-Bpr
and pDG-Mpr, B. subtilis 168 was transformed using these plasmids from E. coli C600 to obtain a kanamycin-resistant strain, 168 (pDG-Vpr), 168 (pDG-AprE), 168 (pDG-Bpr) and 168 (pDG-Mpr).
Preparation of proteins from the supernatant
B. subtilis strains were cultured as described above. The cultures were separated into supernatants and cell debris by centrifugation. For zymography, proteins from the supernatants were solubilized in loading buffer (62.5 mM Tris-HCl [pH 6.8], 2% SDS, 5% 2-mercaptoethanol, 10% glycerol, and 0.05% bromophenol blue). Solubilized samples were analysed by zymography without heat denaturation. Serine protease activities were determined by first adding phenylmethane sulfonyl fluoride (PMSF, 4 mM) to the supernatants from the 8, 25, 50 and 75 h cultures of B. subtilis. The mixtures were incubated for 5 min at room temperature before the loading buffer was added.
Zymography
A separating gel solution (12%, w/v) was prepared in the presence of gelatin (0.1%, w/v). After electrophoresis (at 20 mA for 1.5 h), gels were incubated for 30 min at room temperature in 2.5% Triton X-100 solution. Then the gels were incubated in a zymogram-developing buffer (50 mM Tris-HCl [pH 8.5], 200 mM NaCl, 5 mM CaCl2, and 0.02% Brij35) for 30 min at room temperature, and further incubated in fresh zymogram-developing buffer at 37ºC for 12 h. The gels were stained with Coomassie Brilliant Blue for 1 h, and then destained. The digested bands were visualized as unstained regions in the zymogram gels.
RESULTS AND DISCUSSION
Detection of proteases from the culture supernatant of B. subtilis by gelatin zymography
The supernatant proteins from B. subtilis cultures grown in modified 2xL broth at 8 h (exponential growth phase), 25 h (early stationary phase), 50 h (mid-stationary phase), and 75 h
(late stationary phase) (Fig. 1A) were analyzed by gelatin zymography (Fig. 1B). From the zymogram, the samples with protease activities are distinguishable as clear bands (or zones). In the exponential growth phase (8 h), no protease activity was detected (Fig. 1B, lane 1). However, protease activity increased during the stationary phase, and was highest at 75 h (Fig. 1B, lane 4).
We also examined the zymogram profile of the supernatant from the eight-extracellular-protease-deficient mutant (Dpr8) at 75 h, and found one clear band in the zymogram (Fig. 1C, lane 2). The protease activity was not present in the sample from the aprX mutant at 75 h (Fig. 1C, lane 3). None of the protease activities were present in the sample from the KA8AX strain, in which nine genes (eight extracellular proteases and AprX) were disrupted (Fig. 1C, lane 4). Thus we were able to conclude that the activity from the 75 h culture supernatant of B. subtilis 168 was attributable to AprX as evidenced by a single band observed on the gelatin zymograph (Kodama et al., 2007). To determine the serine protease activities by zymography, PMSF (4 mM) was added to the supernatant from the 8, 25, 50, and 75-h cultures of B. subtilis 168 (Fig. 2). Almost all of the protease activities were decreased. These results indicate that the protease activities from the culture supernatants of B. subtilis, which were detected byzymography, are primarily attributed to the action of serine proteases.
Profile of the protease-deficient mutants by zymography
In order to distinguish each protease, the zymogram pattern of the wild type was compared with the protease-deficient mutants (aprE, bpr, epr, mpr, nprB, nprE, vpr and wprA). Since protease activities were not detected in an 8-h culture as shown in Figures 1 and 2, 40-fold higher volumes of both the supernatants of the wild type and the protease-deficient mutants were applied to the gels for analysis (Fig. 3A). The zymogram showed two active bands except for the vpr mutant (Fig. 3A, lane 8), indicating that both bands were Vpr. When compared with
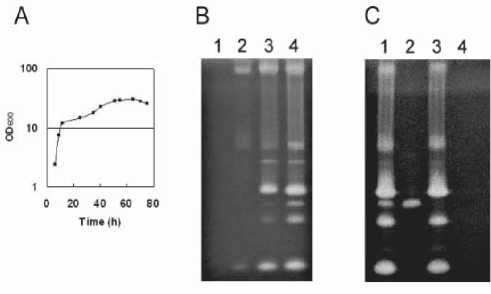
Fig. 1. Zymography of extracellular proteases in B. subtilis strains. (A) Wild type cells were cultured in modified 2xL broth at 30ºC. (B) Protein samples were prepared from the supernatants of the wild type cultured for various incubation times. Lane 1, 8 h culture; lane 2, 25 h; lane 3, 50 h; lane 4, 75 h. (C). Protein samples were prepared from the supernatants of the protease-deficient mutants after a 75 h culture. Lane 1, 168; lane 2, Dpr8; lane 3, AprXdd; lane 4, KA8AX. The samples were analyzed on SDS-12% polyacrylamide gels with 0.1% (w/v) gelatin. Proteins from the culture supernatants (equivalent to 0.1 μl) were applied to each lane for panels B and C.
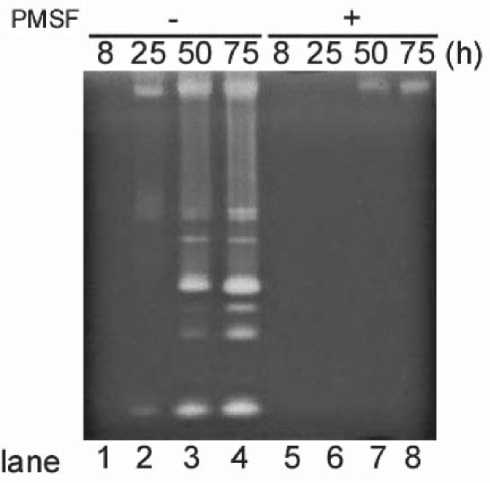
Fig. 2. Zymography analyses of extracellular serine proteases. Culture supernatants from wild type were collected after 8 h (lanes 1 and 5), 25 h (lanes 2 and 6), 50 h (lanes 3 and 7), and 75 h (lanes 4 and 8) of cultivation. Proteins from the culture supernatants (equivalent to 0.1 μl) were applied to each lane. Samples were prepared without 4 mM PMSF (lanes 1-4) and with PMSF (lanes 5-8). The times of harvest of the supernatants are shown at the top.
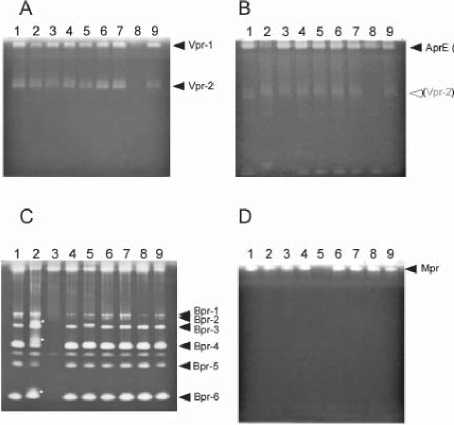
Fig. 3. Zymography of the extracellular protease mutants. Zymography pattern of the wild type was compared with the protease-deficient mutants. Proteins from the supernatants of wild type (lane 1), ∆aprE (lane 2), ∆bpr (lane 3), ∆epr (lane 4), ∆mpr (lane 5), ∆nprB (lane 6), ∆nprE (lane 7), ∆vpr (lane 8), ∆wprA (lane 9). (A) 8-h, (B) 25-h, (C) and (D) 75-h cultures. (D) PMSF (4 mM each) was added to the samples, and then samples of 4 μl (A) and 0.1 μl (B, C and D) of the supernatants were applied to the gels for zymography.
the wild type and the other protease-deficient mutants, the zymogram of the aprE mutant from the 25- and 75 h cultures showed that a protease active band near the top of the gel had greatly decreased (Fig. 3B, lane 2), Therefore, the band was deduced to be AprE. Since one of the deduced Vpr (Vpr-1) bands overlapped with the AprE band, Vpr-1 was concluded to be present in the aprE mutant (Fig. 3B, lane 2). At least 6 major activity bands are lacking in the bpr mutant at 75-h culture (Fig. 3C, lane 3). The results suggest that these activity bands are derived from Bpr. In the aprE mutant, the zymogram profile of Bpr differed from that of wild type (Fig. 3C, lane 2 indicated with asterisks). Thus this suggested that AprE is involved in processing the Bpr protein. The supernatants from the 8, 25, 50, and 75-h cultures were treated with PMSF (final, 4 mM) in order to inactivate the serine proteases (Fig. 2). Almost all of the protease activities were significantly decreased. One specific band was detected in the 50 and 75-h cultures (Fig. 2, lanes 7 and 8). This band was lacking only
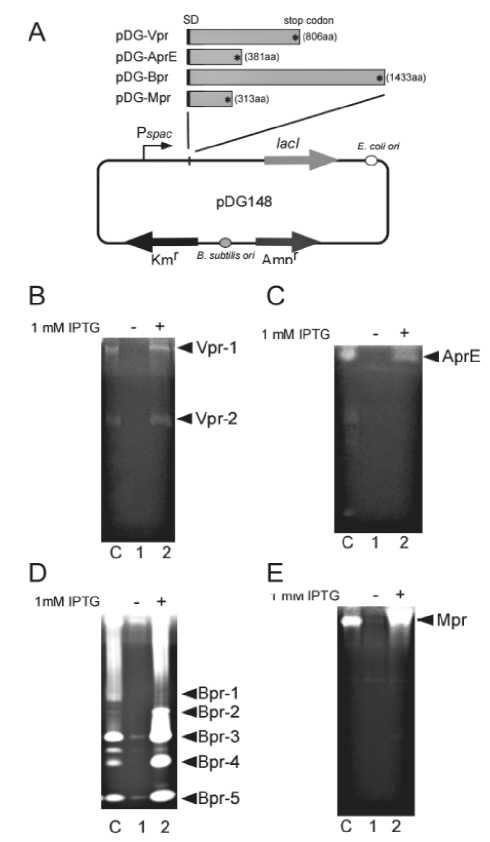
Fig. 4. Detection of extracellular proteases. (A) pDG-Vpr, pDG-AprE, pDG-Bpr and pDG-Mpr were used for expression of Vpr, AprE, Bpr and Mpr proteins in B. subtilis cells. The plasmids contained vpr, aprE, bpr and mpr regions from their SD (Shine-Dalgarno) sequence to the stop codon. To induce the cloned gene by IPTG, the spac promoter was repressed by LacI. Zymography of supernatants from the ∆vpr (pDG-Vpr), ∆aprE (pDG-AprE), ∆bpr (pDG-Bpr), and ∆mpr (pDG-Mpr) strains are shown in panels B, C, D, and E, respectively. Lane C, wild type; lane 1, without IPTG; lane 2, with IPTG. Supernatants from the growth of ∆vpr (pDG-Vpr) (B), ∆aprE (pDG-AprE) (C), ∆bpr (pDG-Bpr) (D), and ∆mpr (pDG-Mpr) (E) strains were collected after 8, 25, 75 and 50-h cultivation, respectively, and supernatant samples of 5, 0.2, 0.1 and 1 μl, respectively, were applied to gels for zymography as described in Materials and Methods.
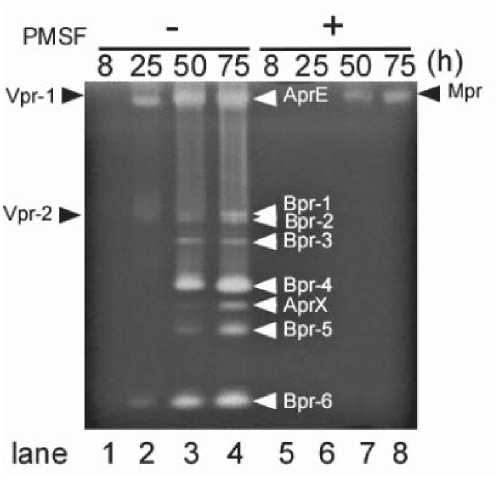
Fig. 5. Identification of extracellular proteases by gelatin zymography. Activities of four extracellular proteases, Vpr, AprE, Bpr and Mpr, were detected by gelatin zymography. Vpr, a minor form of extracellular protease, was detectable at 8 h during the exponential growth phase from the supernatant of B. subtilis culture (Fig. 3A). AprE activity was detected from 25 h of the early stationary phase (Fig. 3B), and Bpr was detected from 50 h of the mid-stationary phase (Fig. 3C). Mpr, a metalloprotease, was detected in the 50 h culture supernatant sample after treatment with PMSF to inactive the serine protease activity (Fig. 3D).
in the mpr mutant (Fig. 3D, lane 5) and thus we concluded that the activity was derived from the metalloprotease, Mpr.
Determination of several proteases in the protease-deficient mutants
We constructed four derivatives of plasmid pDG148 (Fig. 4A). The plasmids contained vpr, aprE, bpr and mpr regions from their SD (Shine-Dalgarno) sequence to the stop codon. These regions were placed downstream of Pspac at SalI and SphI or SalI sites. To induce the cloned gene by IPTG, the spac promoter was repressed by LacI. Vpr was expressed in the vpr mutant carrying pDG-Vpr and then we examined whether the Vpr activities could be detected by zymography (Fig. 4B, lane 2). The result suggested the bands were derived from Vpr. AprE was also detected in the 6-h culture after induction of aprE with IPTG in the aprE mutant carrying pDG-AprE (Fig. 4C
lane 2). Bpr-1, -2, -3, -4, -5 and -6 were detected in the 75-h culture after induction of bpr in the bpr mutant carrying pDG-Bpr (Fig. 4D, lane 2). Mpr was also detected in the 50-h culture after induction of mpr in the mpr mutant harboring pDG-Mpr (Fig. 4E, lane 2). Thus we were able to confirm the detection of Vpr, AprE, Mpr and Bpr by zymography (Fig. 5).
Zymography is a versatile two-stage technique that involves protein separation by electrophoresis, followed by the detection of proteolytic activity (Choi et al., 2004; Kho et al., 2005, Kodama et al., 2007, Park et al., 2002). This technique is routinely used to identify protease activity in polyacrylamide gels under nonreducing conditions. Vpr, a minor form of extracellular protease, was detectable at 8 h during the exponential growth phase from the supernatant of B. subtilis culture (Fig. 3A). AprE activity was detected from 25 h of the early stationary phase (Fig. 3B), and both activities of Mpr and Bpr were shown from 50 h of the mid-stationary phase (Fig. 2, and 3C and D). However, we could not find evidence of other extracellular proteases, Epr, NprB, NprE and WprA with gelatin zymography. In a previous study we reported that the eight extracellular protease genes containing epr, nprB, nprE and wprA are transcribed during the early to late exponential growth phase in modified 2xL broth (Kodama et al., 2007). Although we used skim milk (0.1%) as a substrate for detection of the unidentified proteases by zymography, we could not detect them (data not shown). Zymography of an extracellular protease using a 2 dimensional (D) zymogram gel containing fibrin was previously used to conclude that Vpr has fibrinolytic activity (Kho et al., 2005, Park et al., 2002). Proteome analysis of B. subtilis extracellular proteins by 2D PAGE detected 7 extracellular proteases, AprE, Bpr, Epr, Mpr, NprE, Vpr and WprA, at 1 h after entry into the stationary growth phase cultured with LB medium (Tjalsma et al., 2004). However, Epr, NprE and WprA activities during the mid to late stationary growth phases in modified 2xL broth were not observed in this study. Although
we used gelatin and skim milk as substrates for zymography in this study, the protease activities might be detectable when we use different substrates, such as casein, fibrin, collagen or other proteins.
The active form of the 80-kDa Bpr is produced after the prepro-form of 92-kDa Bpr is processed (Wu et al., 1990). In addition, the 80-kDa protease is gradually processed into various forms with molecular masses of 68, 50, and 48 kDa, respectively (Wu et al., 1990). At least 6 major active bands were shown in the wild type zymogram (Fig. 3C). Thus, the result suggests that Bpr is present as at least 6 active forms extracellularly. We prepared the Bpr protease from ∆bpr (pDG-Bpr) which was grown at 75 h in a medium containing 1 mM IPTG. Bpr-1, -2, -3, -4, -5 and -6 were confirmed by gelatin zymography (Fig. 4D, lane 2). However, Bpr-1 and -2 activities were decreased in the ∆bpr (pDG-Bpr) construct compared with the wild type. On the other hand, the activities of Bpr-3, -4, -5 and -6 were increased. These results suggest that Bpr produced from the ∆bpr (pDG-Bpr) strain underwent significant proteolysis. Bpr from the ∆bpr (pDG-Bpr) construct was produced during the vegetative phase in the presence of 1 mM IPTG therefore the conditions might affect the proteolysis of Bpr. Three separate activities of Bpr derivatives were observed in the aprE- deficient mutant (Fig. 3C, lane 2; indicated with asterisks). This suggested that AprE is involved in processing of the Bpr protein. Previously, it was reported that Vpr was auto-processed to several forms during the desalting step of protein purification. According to our results, Vpr existed as at least 2 active forms in 7 extracellular protease mutants as determined by gelatin zymogram (Fig. 3A). This result is consistent with the previous data that suggested that Vpr adopts several autoprocessed forms.
The four extracellular protease genes of vpr, aprE, mpr and bpr were transcribed at 6, 6, 3 and 12 h of cultivation, respectively (Kodama et al., 2007). The protease activities were observed after a lag of several hours (Figs. 1 to 3). Because
both a signal and “pro” sequence are found in all four extracellular proteases of Vpr, AprE, Bpr and Mpr, these proteases become “active” at a specific time and extracellular location after secretion, folding and propeptide-processing. Active forms of Vpr were observed 2 hours after transcription, since Vpr can easily adopt active forms by auto-processing. We speculated that other proteases containing AprE would be required for processing of Bpr, and this was supported by the fact that Bpr activities were detected a significant time after the transcription of bpr. Mpr may be also processed by other proteases.
The zymography method described herein should be applicable for the rapid detection and identification of extracellular and/or intracellular proteins exhibiting proteolytic activities toward various substrates such as gelatin, casein and other proteins.
ACKNOWLEDGEMENTS
This research was carried out as a part of the Project for the Development of a Technological Infrastructure for Industrial Bioprocesses through R and D on New Industrial Science and Technology Frontiers of the Ministry of Economy, Trade and Industry (METI), and supported by the New Energy and Industrial Technology Development Organization (NEDO).
REFERENCES
Bruckner R, Shoseyov O and Doi RH. 1990. Multiple active forms of a novel serine protease from Bacillus subtilis. Mol. Gen. Genet. 221: 486-490.
Choi NS, Yoo KH, Yoon KS, Maeng PJ and Kim SH. 2004. Nano-scale proteomics approach using two-dimensional fibrin zymography combined with fluorescent SYPRO ruby dye. J. Biochem. Mol. Biol. 37: 298-303.
Kawamura, F and Doi RH. 1984. Construction of a Bacillus subtilis double mutant deficient in extracellular alkaline and neutral
proteases. J. Bacteriol. 160: 442-444.
Kho CW, Park SG, Cho S, Lee DH, Myung PK and Park BC. 2005. Confirmation of Vpr as a fibrinolytic enzyme present in extracellular proteins of Bacillus subtilis. Protein Expr. Purif. 39: 1-7.
Kodama T, Endo K, Ara K, Ozaki K, Kakeshita H, Yamane K and Sekiguchi J. 2007. Effect of the Bacillus subtilis spo0A mutation on cell wall lytic enzymes and extracellular proteases, and prevention of cell lysis. J. Biosci. Bioeng. 103: 13-21.
Kodama T, Endo K, Sawada K, Ara K, Ozaki K, Kakeshita H, Yamane K and Sekiguchi J. 2007. Bacillus subtilis AprX involved in degradation of a heterologous protein during the late stationary growth phase. J. Biosci. Bioeng. 104: 135-143.
Kunst F. 1997. The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature 390: 249-256.
Margot P. and Karamata D. 1996. The wprA gene of Bacillus subtilis 168, expressed during exponential growth, encodes a cell-wall-associated protease. Microbiology 142: 3437-3444.
Park SG, Kho CW, Cho S, Lee DH, Kim SH and Park BC. 2002. A functional proteomic analysis of secreted fibrinolytic enzymes from Bacillus subtilis 168 using a combined method of two-dimensional gel electrophoresis and zymography. Proteomics 2: 206-211.
Rufo GA Jr, Sullivan BJ, Sloma A and Pero J. 1990. Isolation and characterization of a novel extracellular metalloprotease from Bacillus subtilis. J. Bacteriol. 172: 1019-1023.
Sloma A, Ally A, Ally D and Pero J. 1988. Gene encoding a minor extracellular protease in Bacillus subtilis. J. Bacteriol. 170: 5557-5563.
Sloma A, Rudolph CF, Rufo GA Jr, Sullivan BJ, Theriault KA, Ally D and Pero J. 1990. Gene encoding a novel extracellular metalloprotease in Bacillus subtilis. J. Bacteriol. 172: 1024-1029.
Sloma A, Rufo GA Jr, Rudolph CF, Sullivan BJ, Theriault KA and Pero J. 1990. Bacillopeptidase F of Bacillus subtilis: purification of the protein and cloning of the gene. J. Bacteriol. 172:1470-1477. Erratum in: 1990. J. Bacteriol. 172: 55205521.
Sloma A, Rufo GA Jr, Theriault KA, Dwyer M, Wilson SW and Pero J. 1991. Cloning and characterization of the gene for an additional extracellular serine protease of Bacillus subtilis. J. Bacteriol. 173: 6889-6895.
Stahl ML and Ferrari E. 1984. Replacement of the Bacillus subtilis subtilisin structural gene with an in vitro-derived deletion mutation. J. Bacteriol. 158: 411-418.
Stragier P, Bonamy C and Karmazyn-Campelli C. 1988. Processing of a sporulation sigma factor in Bacillus subtilis: How morphological structure could control gene expression. Cell 52: 697–704.
Tjalsma H. 2004. Proteomics of protein secretion by Bacillus subtilis: separating the “secrets” of the secretome. Microbiol Mol Biol Rev. 68: 207-233.
Tran L, Wu XC and Wong SL. 1991. Cloning and expression of a novel protease gene encoding an extracellular neutral protease from Bacillus subtilis. J. Bacteriol. 173: 63646372.
Wong SL, Price CW, Goldfarb DS and Doi RH. 1984. The subtilisin E gene of Bacillus subtilis is transcribed from a σ37 promoter in vivo. Proc. Natl. Acad. Sci. USA 81: 11841188.
Wu XC, Nathoo S, Pang AS, Carne T and Wong SL. 1990. Cloning, genetic organization, and characterization of a structural gene encoding bacillopeptidase F from Bacillus subtilis. J. Biol. Chem. 265: 6845-6850.
Yang MY, Ferrari E and Henner DJ. 1984. Cloning of the neutral protease gene of Bacillus subtilis and the use of the cloned gene to create an in vitro-derived deletion mutation. J. Bacteriol. 160: 15-21.
66 • udayana university biosciences and biotechnology forum
Discussion and feedback