BETA-AMYLOID AS PATHOGENESIS OF ALZHEIMER DISEASE
on
BETA-AMYLOID SEBAGAI PATOGENESIS PADA PENYAKIT ALZHEIMER
Kadek Ary Mahendri Pattni
Bagian / SMF Psikiatri Rumah Sakit Umum Pusat Sanglah / Fakultas Kedokteran Universitas Udayana
ABSTRAK
Penyakit Alzheimer sering menyebabkan penurunan kognitif pada populasi usia lanjut. Insiden terjadinya penyakit Alzheimer meningkat sesuai umur antara 0,3% - 0,6% terjadi pada usia 65 – 69 tahun dan 5,3% - 7,5% terjadi pada usia 85 – 90 tahun. Meskipun demikian Alzheimer juga dapat terjadi pada usia kurang dari 65 tahun dikenal dengan Alzheimer onset dini yang disebabkan oleh peningkatan agregasi dari Beta-Amyloid (Aβ) dari hasil mutasi amyloid precursor ptotein (APP). Akumulasi substansi inilah yang nantinya dapat memicu kaskade hilangnya sejumlah sinaps di otak sehingga menimbulkan Dementia tipe Alzheimer.
Kata kunci : Alzheimer, Beta-Amyloid
BETA-AMYLOID AS PATHOGENESIS OF ALZHEIMER DISEASE
ABSTRACT
Alzheimer disease is often causing cognitive decline in elder population. The incidence of this disease is increasing by the age, about 0,3% to 0,6% it affects individuals aged 65 to 69 years, and 5,3% - 7,5% affects individuals aged 65 to 69 years. Alzheimer also affects individuals aged less then 65 years and known as early onset of Alzheimer disease that is caused by increasing of Beta-Amyloid aggregation from the mutation of amyloid precursor protein (APP). From the accumulation of this substance may trigger the cascade lost of some synaps in the brain and arising Alzheimer type of Dementia.
Keywords : Alzheimer, Beta-Amyloid
PENDAHULUAN
Penyakit Alzheimer merupakan penyebab tersering timbulnya dementia dan menyebabkan gangguan kognitif pada populasi usia lanjut.1,2 Dementia pada penyakit Alzheimer memiliki onset yang gradual dan adanya penurunan kognitif secara berkelanjutan termasuk gangguan memori dan adanya satu atau lebih aphasia (gangguan bahasa), apraxia (gangguan fumgsi motorik), agnosia (gangguan fungsi sensoris), dan gangguan fungsi eksekutif seperti ketidakmampuan perencanaan, pengorganisasian, serta melakukan aktivitas normal.3,4
Insiden penyakit Alzheimer dibagi menjadi 2 kelompok yaitu kelompok yang menderita pada usia 65 tahun kebawah (onset dini) dan kelompok yang menderita pada usia 65 tahun keatas (onset lanjut).3 Insiden terjadinya penyakit Alzheimer meningkat sesuai umur antara 0,3% - 0,6% terjadi pada usia 65 – 69 tahun dan 5,3% - 7,5% terjadi pada usia 85 – 90 tahun.5
Terjadinya penyakit Alzheimer onset lanjut dihubungkan dengan adanya apolipoprotein E. Sedangkan penyakit Alzheimer onset dini tipe familial dihubungkan dengan 3 gen yang mengalami mutasi yaitu amyloid precursor protein (APP), presenilin-1 (PS1), dan presenilin-2 (PS2).1,2,3 Dimana mutasi ini terjadi dalam produksi yang berlebih dan/atau adanya peningkatan agregasi dari Beta-Amyloid (Aβ).6,7 Hal ini menjadi dasar penulisan makalah ini. Penulis akan mencoba menjelaskan lebih lanjut pada bab pembahasan.
PENYAKIT ALZHEIMER DAN KAITANNYA TERHADAP BETA-AMYLOID
Pada penyakit Alzheimer ditemukan karakteristik neuropatologikal seperti hilangnya neuronal selektif dan sinap, adanya plak neuritis yang mengandung peptida Aβ dan neurofibrillary tangles (NFTs) yang membentuk hiperfosforilasi dari protein tau. Plak neuritik yang terjadi merupakan lesi ekstraseluler yang tersusun atas inti sentral dari agregasi Aβ peptida yang dikelilingi oleh neurit distropi, mikroglial yang teraktivasi, dan atrosit reaktif. Sedangkan NFTs merupakan buntalan filamen di dalam sitoplasma sel saraf yang mengelilingi sel saraf. Gambar 1 akan memberikan gambaran mengenai perubahan 2
tingkat seluler tersebut.2
Deposisi Aβ pada otak merupakan salah satu implikasi dari patogenesis penyakit Alzheimer. Akumulasi Aβ (khususnya Aβ42 peptida) pada otak merupakan inisiasi terjadinya disfungsi neuron, neurodegenerasi, dan dementia.2,9,10 Mutasi gen APP pada kromosom 21, PS1 pada kromosom 14, dan PS2 pada kromosom 1 mengarah pada early-onset penyakit Alzheimer tipe familial yang terjadi dalam produksi berlebihan dan/atau peningkatan agregasi dari Aβ. Beta-Amyloid merupakan produk fisiologi normal dari APP dan merupakan komponen soluble dari plasma dan cairan cerebrospinal.9
Dalam pembentukan Aβ, APP dipecah oleh tiga enzim yaitu α-, β-, dan γ-secretase. Pemecahan APP oleh β-secretase kemudian oleh γ-secretase menghasilkan Aβ sedangkan bila dipecah oleh α-secretase akan menghasilkan peptida yang bersifat nontoxic.2,3 Mutasi ganda pada APP mengarah pada peningkatan Aβ akibat pemecahan APP oleh β-secretase yang meningkat. Beta-Amyloid peptida merupakan komponen protein utama pada plak neuritik yang merupakan karakteristik dari penyakit Alzheimer.2
Beta-Amyloid terkadang memulai aksi toksik sebelum terbentuknya fibril. Peningkatan derajat Aβ soluble dan bukan plak Aβ berhubungan dengan disfungsi kognitif pada penyakit Alzheimer. Adanya gangguan kognitif pada individu yang menderita penyakit Alzheimer sangat kuat dihubungkan dengan hilangnya sinap yang melewati region kortikal otak.10,11 Self-agregation dari Aβ menjadi oligomer soluble low-n merupakan penyebab utama sinaptoksisitas pada penyakit Alzheimer. Terdapat dua varian terminal karboksil dari Aβ yaitu Aβ40 yang merupakan sekret spesies utama dari sel kultur dan terdapat pada cairan cerebrospinal sedangkan Aβ42 merupakan komponen utama amyloid yang berdeposit di otak pada penyakit Alzheimer. Peningkatan Aβ42 lebih sering mengalami agregasi dan membentuk fibril. Neurotoksik yang dihasilkan oleh agregasi Aβ menghasilkan beberapa mekanisme, seperti adanya akumulasi radikal bebas, disregulasi dari homeostatis kalsium, respon inflamasi, dan adanya aktivasi dari beberapa signaling pathway.9
Akumulasi Radikal Bebas
Pada penyakit Alzheimer terjadi neurodegenerasi pada otak dan adanya stress oksidatif yang dihubungkan dengan adanya peningkatan deposit Aβ. Walaupun sebenarnya mekanisme pastinya belum sepenuhnya dimengerti, Aβ yang kontak atau masuk ke neuronal dan glial membran bilayer menghasilkan radikal bebas oxygen-dependent yang kemudian menyebabkan peroksidasi lipid dan oksidasi protein. Beta-Amyloid menyebabkan akumulasi H2O2 pada kultur neuron hippokampus dan pada kultur neuroblastoma. Oligomer Aβ menyebabkan lepasnya lipid dari membran neuron mengakibatkan gangguan homeostatis lipid neuron dan hilangnya fungsi neuron. Hilangnya integritas membran
akibat radikal bebas yang dihasilkan oleh Aβ mengarah pada disfungsi selular, seperti inhibisi dari ion-motive ATPase, inhibisi sistem pengambilan glutamat dari sel glial Na+-dependent sebagai konsekuensi dari eksitasi neuron reseptor N-methyl-D-aspartate (NMDA) yang dapat menyebabkan hilangnya sinap pada neuron, hilangnya homeostatis kalsium, hilangnya fungsi transporter protein, gangguan signaling pathway, dan aktivasi faktor transkripsi nuklear serta jalur apoptosis.3,9
Gangguan Homeostatis Kalsium
Kalsium merupakan salah satu messengers intraselular yang penting pada otak karena fungsinya sebagai perkembangan neuron, transmisi sinap, kekenyalan otak, dan meregulasi jalur metabolik yang bervariasi. Neurotoksisitas akibat kalsium yang dimediasi oleh Aβ peptida terjadi karena Aβ peptida dapat meningkatkan influx kalsium melalui voltage-gate channel kalsium, membentuk channel ion yang kationnya bersifat selektif setelah Aβ peptida bergabung dalam membran selular, mengurangi blockade magnesium dari reseptor N-methyl-D-aspartate (NMDA) agar dapat meningkatkan influx Ca2+, dan menghambat channel K+ dan pertukaran Na+/Ca2+.3,9,10
Respon Inflamasi
Asosiasi awal dari sel mikrogial yang teraktivasi dan reaktif astrosit pada plak neuritis dan adanya penanda inflamasi mengindikasikan adanya inflamasi kronik pada penyakit Alzheimer. Pada penderita penyakit Alzheimer terjadi peningkatan aktivasi imun dan/atau aktivitas inflamasi pada otak penderita dibandingkan dengan yang tidak menderita penyakit tersebut. Adanya proses neuroinflamasi yang terjadi secara terus-menerus disertai aktivasi sel glial merupakan salah satu pathogenesis terjadinya penyakit Alzheimer. Aktivasi
mikroglial yang diinduksi oleh adanya Aβ berperan sebagai trigger dalam jalur komplement klasik dan sebagai trigger dalam produksi sitokin proinflamasi yang bervariasi. Protein komplement merupakan komponen integral plak amyloid dan adanya vaskularisasi cerebral amyloid pada otak penderita penyakit Alzheimer. Hal ini dapat ditemukan pada tahap awal deposisi amyloid dan aktivasinya bertepatan dengan adanya ekspresi klinis dementia pada penyakit Alzheimer.8,9,10
Sebuah penelitian juga menunjukkan adanya aktivasi dari jalur mitogen-activated protein kinase (MAPK) sebagai respon adanya fibril Aβ setelah terjadinya sinyal inflamasi tyrosine kinase-dependent pada mikroglial. Sel mikroglial yang terpapar Aβ akan mengsekresi mediator inflamasi, seperti sitokin, kemokin, growth factor, komplement, dan intermediate reaktive. Adanya paparan preagregasi Aβ42 menyebabkan peningkatan dari prointerleukin-1β, interleukin-6, tumor necrosis factor-β, monocyte chemoattractant protein-1, macrophage inflammatory peptide-1β, IL-8, dan macrophage colony-stimulating factor. Respon inflamasi yang diinduksi oleh Aβ merupakan mediator penting terjadinya cedera neuron pada penyakit Alzheimer.2,8
Aktivasi Signaling Pathway
Beta-Amyloid telah diketahui dapat mengaktifkan signaling pathway, seperti MAPK pathway dan juga Aβ berperan dalam terjadinya hiperposphorilasi dari microtubule-associated protein τ (MAPT). Produksi yang berlebihan dan adanya agregasi dari MAPT dapat menyebabkan hilangnya sinap pada neuron akibat pengaruh Aβ.9
Hipotesis Alur Amyloid
Hipotesis alur Amyloid menunjukkan bagaimana Amyloid dapat menimbulkan adanya dementia pada penyakit Alzheimer. Diawali dengan terjadinya kegagalan clearance mechanisms dari Aβ atau overproduksi Aβ42, akibat kegagalan clearance terjadi akumulasi dan oligomerisasi dari Aβ42 di limbik dan berhubungan dengan korteks. Akumulasi ini berlangsung terus-menerus secara bertahap sebagai plak. Plak yang terjadi mengaktivasi mikroglial dan atrosit sebagi respon inflamasi. Kemudian terjadi perubahan homeostatis neuron, dan terjadi oxidative injury yang mengakibatkan perubahan aktivitas kinase ataupun phospat. Perubahan ini menyebabkan terjadinya hiperposporilasi dari protein tau yang akan membentuk Neurofibrillary tangles. Disfungsi sinap atau neuron dan hilangnya neuron selektif diikuti dengan adanya penurunan neurotransmitter merupakan perubahan yang terjadi akibat Neurofibrillary tangles. Adanya Neurofibrillary tangles yang mengakibatkan hilangnya sinap pada saraf dapat menyebabkan timbulnya dementia pada penyakit Alzheimer.
RINGKASAN
Penyakit Alzheimer merupakan penyebab tersering timbulnya dementia dan menyebabkan gangguan kognitif pada populasi usia lanjut. Dementia pada penyakit Alzheimer memiliki onset gradual dengan penurunan kognitif yang berkelanjutan. Kelainan yang ditimbulkan meliputi gangguan memori, berbahasa, fungsi motorik, fungsi sensoris, dan gangguan fungsi eksekutif.
Penyakit Alzheimer dikarakteristikan dengan adanya neuropathologikal seperti hilangnya neuronal selektif dan disfungsi sinap, adanya plak neuritis yang mengandung Aβ
peptida dan neurofibrillary tangles (NFTs). Peningkatan Aβ42 lebih merupakan pathogenesis penyakit Alzheimer, yang diawali dengan kegagalan clearance mechanisms dari Aβ / overproduksi Aβ42, hingga menimbulkan akumulasi dan oligomerisasi dari Aβ42 di limbik yang berhubungan dengan korteks. Akumulasi ini berlangsung terus-menerus secara bertahap sebagai plak. Plak yang terjadi mengaktivasi mikroglial dan atrosit sebagi respon inflamasi. Kemudian terjadi perubahan homeostatis neuron, dan terjadi oxidative injury yang mengakibatkan perubahan aktivitas kinase ataupun fosfat.
Perubahan aktivitas kinase ataupun fosfat menyebabkan terjadinya hiperposporilasi dari protein tau yang akan membentuk Neurofibrillary tangles. Disfungsi sinap atau neuron dan hilangnya neuron selektif diikuti dengan adanya penurunan neurotransmitter merupakan perubahan yang terjadi akibat Neurofibrillary tangles. Adanya Neurofibrillary tangles yang mengakibatkan hilangnya sinap pada saraf dapat menyebabkan timbulnya dementia pada penyakit Alzheimer.
DAFTAR PUSTAKA
-
1. Jayadev S, Steinbart EJ, Chi YY, Kukull WA, Schellenberg GD, Bird TD. Conjugal alzheimer disease: risk in children when both parents have alzheimer disease. Arch Neurol. 2008; 65(3): 373-378.
-
2. Klafki HW, Staufenbiel M, Kornhuber J, Wiltfang J. Therapeutic approaches to alzheimer’s disease. Brain. 2006; 129: 2840-2855.
-
3. Richardsz SS, Sweet RA. Dementia. In: Sadock BJ, Sadock VA, Ruiz P. Comprehensive text book of psychiatry volume 1. 9th ed. Philadelphia: Lippincott Williams and Wilkins. 2009; 1176-1185.
-
4. Salloway S, Correia S. Alzheimer disease: time to improve its diagnosis and treatment. Cleveland Clinic Journal of Medicine. 2009; 1: 49-58.
-
5. Basset SS, Yousem DM, Cristinzio C, Kusevic I, Yassa MA, Caffo BS, et al. Familial risk for alzheimer’s disease alters: fMRI activation patterns. Brain. 2006; 129: 12291239.
-
6. Rapport BJM. Does this patient have alzheimer disease?: diagnosing and treating dementia. Cleveland Clinic Journal of Medicine. 2003; 70(9): 762-776.
-
7. Emilien G, Beyreuther K, Masters CL, Maloteaux JM. Prospects for pharmacological intervention in alzheimer disease. Arch Neurol. 2000; 57: 454-459.
-
8. Prasad KN, Cole WC, Prasad CK. Risk factor for alzheimer disease: role of multiple antioxidant, non-steroidal anti-inflammatory and cholinergic agent alone or in combination in prevention and treatment. J Am Coll Nutr. 2002; 21(6): 506-522.
-
9. Suh YH, Checler F. Amyloid precursor protein, presenilins, and α-synuclein: molecular pathogenesis and pharmacological application in alzheimer disease. Pharmacol Rev. 2002; 54: 469-525.
-
10. Masters CL, Beyreuther K. Alzheimer’s centennial legacy: prospects for rational therapeutic intervention targeting the Aβ amyloid pathway. Brain. 2006; 129: 28232839.
-
11. Pike KE, Savage G, Villemagne VL, Ng S, Moss SA, Maruff P, et al. β-amyloid imaging and memory in non-demented individual: evidence for preclinical alzheimer’s disease. Brain. 2007; 130: 2837-2844.
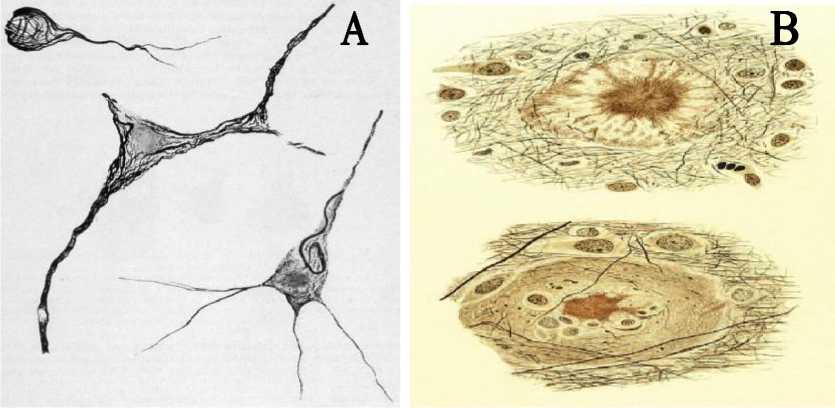
Gambar 1. (A) Gambaran perubahan neurofibrilar pada sitoplasma sel saraf. (B) Plak neuritik dengan inti sentral Amyloid dan dihubungkan dengan reaksi sel glial.
Sumber: Alzheimer’s Centennial Legacy: Prospects for Rational Therapeutic Intervention Targeting The Aβ Amyloid Pathway. Oxford University Press. 2006
11
Discussion and feedback