THE EFFECT OF THE D185 MUTATION ON THE STABILITY AND FUNCTIONALITY OF RUBISCO LIKE PROTEIN (RLP) FROM CHROMOHALOBACTER SALEXIGEN BKL 5
on
JURNAL KIMIA (JOURNAL OF CHEMISTRY) 18 (1), JANUARI 2024
DOI: https://doi.org/10.24843/JCHEM.2024.v18.i01.p01
p-ISSN 1907-9850
e-ISSN 2599-2740
EFFECT OF THE D185 MUTATION ON THE STABILITY AND FUNCTIONALITY OF RUBISCO LIKE PROTEIN (RLP) FROM CHROMOHALOBACTER SALEXIGEN BKL 5
I. Sudarmanto 1,2, M. S. Rohman2,3*, W. T. Artama2,4
-
1Chemistry Study Program, Department of Sciences, Institute Technology of Sumatera, Jl Terusan Ryacudu, WayHui, Lampung Selatan, 35365, Lampung
-
2Post Graduate Program of Biotechnology, Post Graduate Program, University of Gadjah Mada, Jl
Teknika Utara, Catur Tunggal, Sleman, 55281, Yogyakarta
-
3Department of Agricultural Microbiology, Agriculture Faculty, University of Gadjah Mada, Karang Malang, Catur Tunggal, Sleman, 55281, Yogyakarta
-
4Veterinary Medicine Faculty, University of Gadjah Mada, Jl Fauna No 2, Karang Gayam, Catur
Tunggal, Sleman, 55281, Yogyakarta
*Email: saifur@ugm.ac.id
ABSTRACT
In the case of Rubisco Like Protein (RLP) Chromohalobacter salexigen BKL 5 (RLP CS), which is halophilic, the presence of salt bridges (SB) is not too numerous but efficient enough to maintain stability in high salt stress. This study was to determine the effect of mutations SB at position 185 (D185/WT) on the stability and activity of RLP CS through an in-silico approach by replacing aspartic acid with glutamic acid and alanine (D185E and D185A). The methods used were Molecular Dynamics Simulations (MDS) and Molecular Docking (MD). The MDS was used to study molecular characteristics at the atomic level, while MD was for the interaction patterns of proteins and ligands. The results of the MDS analysis carried out at 10ns showed that the mutation at position 185 with alanine and glutamic acid changed the size/dimensional of RLP CS and affected its overall geometric structure. Interestingly, even though the structure has changed, the activity of the protein remains relatively constant, which is indicated by the results of MD, and has a relatively similar binding energy value of around -6.3 Kcal/mol.
Keywords: Chromohalobacter salexigen BKL 5, molecular docking, molecular dynamic simulation, mutation, saltbridge
INTRODUCTION
Saltbridge (SB) is defined as the electrostatic interaction between two oppositely charged groups: anionic carboxylates from glutamic acid (E) or aspartic acid (D), and cationic ammonium from arginine (R) or lysine (K) (Bosshard et al., 2004; Musafia et al., 1995). Salt bridge interactions in proteins can provide structural and functional conformation and contribute to protein recognition, degradation, catalysis, and stability. (Albeck et al., 2000; Kumar & Nussinov, 2002).
Mutation analysis is a useful procedure for estimating the contribution of several factors to the conformational stability of a protein. The effects of mutations on protein stability differ from site to site, depending on their environment within the structure (Takano et al., 1995; Takano et al., 2000). One approach that is often used to predict the effect of mutations on proteins is MDS. Molecular dynamics (MD) simulations can predict how individual atoms in
a protein or other molecular system will move over time, based on general models of physics governing interatomic interactions (Karplus and McCammon, 2002; Rácz et al., 2022). These simulations can capture a wide variety of important biomolecular processes, including conformational changes, ligand binding, and protein folding, revealing the positions of all atoms at femtosecond temporal resolution. Such simulations can also predict how biomolecules will respond at the atomic level to perturbations such as mutations, phosphorylation, protonation, or the addition or removal of ligands.
Simulations can also predict the behavior of proteins and other biomolecules in full atomic detail and at very fine temporal resolution. Substantial improvements in the speed, accuracy, and accessibility of simulations, together with the proliferation of experimental structural data, have increased the attractiveness of biomolecular simulations for experimentalists. Simulations have proven
valuable in deciphering the functional mechanisms of proteins and other biomolecules in uncovering the structural basis of disease and in the design and optimization of small molecules, peptides and proteins (Hollingsworth & Dror, 2018).
The results of previous observations showed that the saltbridge in halophilic RLP only has half of its homologous RLP saltbridge which is mesophilic (unpublished). A more detailed observation of the RLP CS saltbridge shows that the saltbridge at position D185 is unique because it has the most interactions and is closest to the active site of the protein which is thought to have a major contribution in maintaining the stability (conformation) of RLP CS to function optimally. This study was conducted to determine how much influence the saltbridge has on the function and stability of RLP cs. Tests were carried out through the process of mutation of aspartic acid with other residues (glutamic acid and alanine) and analyzed using a computational approach, namely Molecular dynamic simulation and molecular docking methods. It is hoped that the results of this study can provide an overview of how the saltbridge influences the stability and activity of RLP CS.
MATERIAL AND METHODS
Material and Tool
The 3D structure of RLP CS in the form of PDB files, called as Wild Type (WT). Protein sequences in FASTA format containing mutants at position 185, hereinafter referred to as D185E (asp mutation to glutamic acid) and D185A (aspartic acid mutation to alanine). The computational analysis was operated on ACER Nitro with i7 processor, 32 GB RAM, Windows 10, GPU GTX 1060 6 GB and 64-bit operating system
Method
The first step is identification and visualization of the areas/residues of amino acids to be mutated using the Chimera tool, followed by the following steps.
Homologation of mutational 3D structures
Mutations were carried out on the aspartic acid amino acid residue at position 185 (D185/WT) by replacing it with glutamic acid (D185E) and alanine (D185A). The selection of these two types of residues was based on
previous study which showed that RLP CS has significant differences in glutamic acid and alanine compared to its non-halophilic homologs (unpublished). Sequences containing mutant residues were homologated with AlphaFold to obtain a 3D structure in the form of PDB files (Miyazono & Tanokura, 2022). The selected structures were validated with ERRAT and SAVE (Messaoudi et al., 2013).
The results of the homologation were analyzed for energy in the ground state with the SPDV Tool (Guex & Peitsch, 1997) then superimposed with RLP WT to find out the difference in geometry in the form of RMSD using Chimera (Pettersen et al., 2004).
Molecular Dynamic Simulation
Molecular dynamics simulations can be used as a procedure to study molecular characteristics at the atomic level, such as protein conformation changes that occur. In this study protein molecular dynamics (MDS) simulations were carried out using the Gromacs-based method performed on the WebGro server (Oostenbrink et al., 2004; Abraham et al., 2015).
Proteins were prepared using the GROMOS96 43a1 force field. SPC was chosen as a model solvent (a triclinic water box with a size of 50 × 75 × 70 Å) for protein complexes. This system is neutralized by adding sodium or chlorine ions based on the total charge. To minimize the system before MD, the steepest descent (5000 steps) algorithm was applied. MD simulations were carried out in the presence of 0.15 M NaCl using constant temperature (300 K) and pressure (1.0 bar). The approximate number of frames per simulation is 1000. The simulation time is set to 10 ns. The parameters analyzed were RMSD, RMSF, Rg, SASA and the number of hydrogen bonds formed. Graph preparation and visualization using Originlab 2019 and GraPhadPrism 8.
Molecular Docking
To predict the interaction patterns of proteins and ligands, a molecular docking process was carried out. In this study the method used was Autodock Vina and the MGL Tool. The ligand used was RUBP whose structure was obtained from a web-based portal (Pubchem) in SDF format. The protein used is the PDB structure of the RLP WT file and the mutants prepared using the MGL Tool (Morris et al., 2009). All polar hydrogens were removed
from the ligand and a Gasteiger type charge was added. Thereafter the receptor files were prepared by adding hydrogen and a Kollman charge. Then all files are converted to pdbqt format for docking. The grid is arranged according to the structure in such a way as to cover the active site area of the RLP protein. After preparation of the ligand and receptor, docking was carried out using Autodock Vina (Walker & Causgrove, 2009). The best docking position (the most negative binding energy value) was chosen for visualization. Visualization of docking results using LigPlot.
RESULT AND DISCUSSION
Visualization of SB at position 185 and Homologation of Mutant Structures
Previous studies revealed that the aspartic acid (asp/D) residue at position 185 is unique because it has interactions with several other residues to produce a relatively large amount of saltbridge (SB) compared to other residues that make up SB. Visualization of SB
at position 185 is shown in Figure 1. In the figure it can be seen that aspartic acid at position 185 binds to arginine (R) at 146,221 and 394 which forms a salt bridge which is important to maintain conformation, especially the active site area.
The results of the homologation with AlphaFold which have been validated (data not shown) are then analyzed for energy and compared with WT which shows that the two mutants are geometrically different (Table 1). This is consistent with initial predictions that changing or even removing the saltbridge will affect the stability of the RLP CS (WT) protein, considering that the saltbridge is very important to maintain the structure of a protein. The small RMSD value of the superimposed results on the two mutants indicates that the two mutants are geometrically smaller than the WT structure of RLP CS and this is reinforced by the energy value which also changes. This indicates that mutations in the saltbridge area will reduce stability.
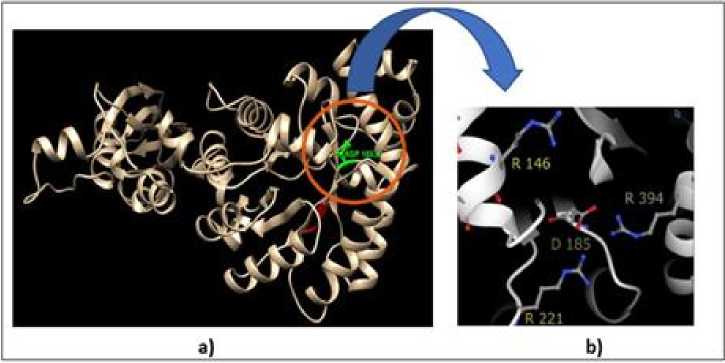
Figure 1. Position of Aspartic Acid at Number 185 (A) and the Salt-bridge Interaction Formed (B)
Table 1. Characteristics of WT and Mutation Results
|
Protein RMSD |
ΔG Binding affinity Volume active site |
|
Compare to WT |
KJ/mol KCal/mol (SA) Å3 |
|
WT 0 |
22.444.898 -6,3 376.486 |
|
D185E 0,403 |
20.364.660 -6,2 364.192 |
|
D185A 0,685 |
20.182.055 -6,3 372.971 |
Molecular Dynamics
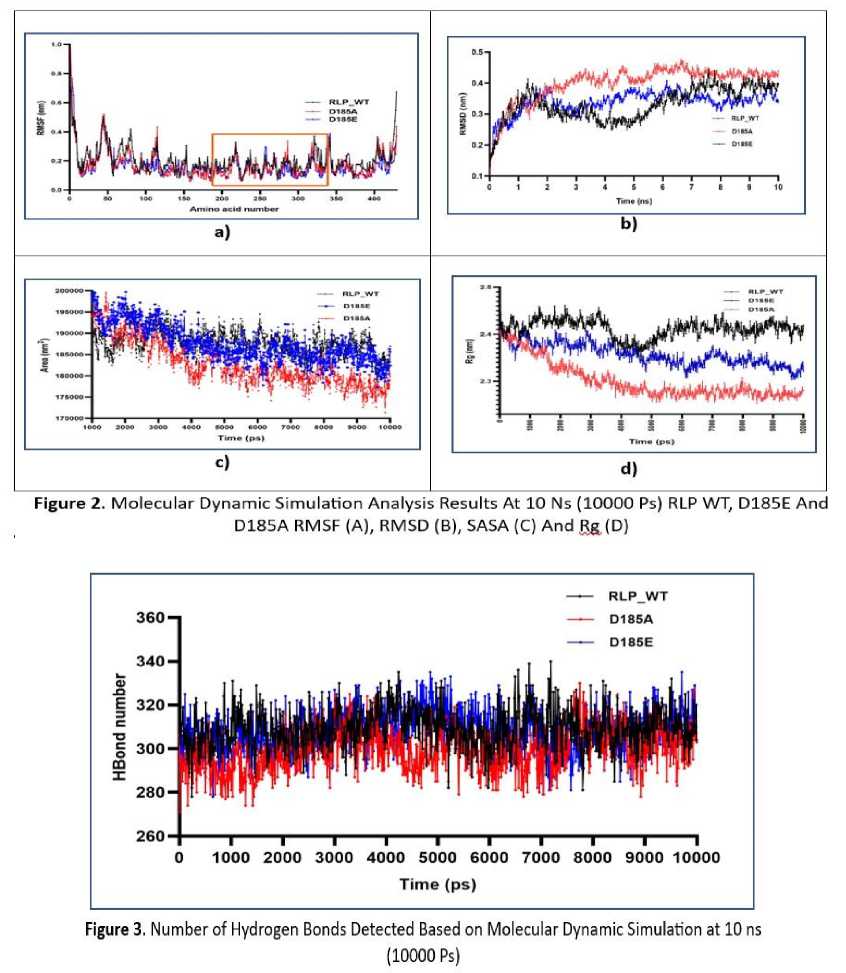
RMSD is a parameter commonly used to determine the stability of a system (protein). RMSD can describe the conformational and geometric changes of a protein. The greater the RMSD value, the more unstable the protein will be and vice versa. The MDS analysis shows that the RMSD value of the mutant is greater than that of WT which illustrates that the conformation of the mutant protein changes and becomes unstable compared to WT (Figure 2b). This happens because there is no salt bridge that allows the protein to lose its stability. Although the D185E mutant still has a saltbridge, its RMSD value is still greater than that of WT, indicating that aspartic acid is the best residue that can maintain the stability of the RLP protein in a halophilic state.
The conformational geometry of WT and mutant proteins can be observed by studying the radius of gyration (Rg), SASA, and hydrogen bond analysis (Shukla et al., 2018). The Rg of a protein describes the size of the protein globally, the higher the value, the more open the protein tends to be and vice versa. A small Rg value indicates that the protein is in a compact state while a large Rg value indicates that the protein has a smaller folding structure. In this study, both mutants showed smaller Rg values than their WT, especially the D185A mutant (Figure 2d). This shows that D185A has smaller dimensions than WT and tends to be more folded than WT and D185E. This fact was further confirmed by observing the SASA parameters and the number of hydrogen bonds formed.
SASA is defined as protein accessibility to water (solvent). Lower SASA value determines a compact structure and vice versa. SASA can also describe folding patterns in proteins, where a small SASA value indicates that there is high folding. In this case, the WT and mutant proteins show different SASA patterns, the D185A area has the smallest value compared to D185E and WT. (Figure 2c). This occurs because the aspartic acid mutation with alanine (D185A) will remove the saltbridge so that the protein structure will fold in response to
hydrophobic amino acids in protein to water (solvent). At D185E the SASA value will also decrease because even though the saltbridge is still formed, the glutamic acid side chain is longer so that it will affect the distance the saltbridge is formed (longer).
The number of hydrogen bonds in a protein describes the atomic level interactions. It also validates the compactness or shape of the structure. The compact structure of proteins is generally advantageous for intramolecular hydrogen bond interactions. In order to predict how the mutation affects the structural stability, the intramolecular hydrogen bonds of WT and the mutant are shown in Figure 3. It was observed that the WT protein and the mutant protein exhibited different numbers of hydrogen bonds. In general, D185A has fewer hydrogen bonds than WT and D185E. This strengthens the notion that the structure of D185A is more compact because fewer interactions involving hydrogen bonds are formed in its molecule.
The results of the comparison of Rg and SASA and the number of hydrogen bonds in the molecule against WT and mutants strengthen the notion that the mutation in D185 affects the conformation and geometry of RLP globally.
RMSF is a parameter that can describe the mobility/flexibility of individual amino acid residues. Rigid structures such as helices and sheets will have a high RMSF value while loops and turns will have a low RMSF value. Calculation of the RMSF value in the RLP WT and mutants shows that the RMSF value of the mutant has a lower value compared to the RLP WT (Figure 2a). This fact strengthens the notion that mutant RLP has higher cohesiveness than WT which causes the amino acid residues to be more bound. Observation of the residues in the active site area (190-320) shows that the residue in that area has almost no difference between WT and its mutants which indicates that the active site area is conserved and stable.
Molecular Docking
Molecular docking is performed to predict changes in active site residues after mutation by observing the interactions between the ligand and amino acid residues in the active site area and measuring the binding energy formed (Meng et al., 2011; Al-Karmalawy et al., 2021). The ligand used was Ribulose 1.5 Biphosphate as a substrate for the Rubsico Like Protein from Chromohalobacter salexigen BKL 5 (RLP CS) protein analogue.
The docking results showed that there was no change in the interaction between the ligand and the active site of the protein which was characterized by similar binding energy values (Table 1). This shows that the mutation
does not cause changes in protein activity. This fact occurs because the active site region which is responsible for protein activity has not changed which is confirmed by the results of analysis using CastP 3.0 (Tian et al., 2018) which indicates that the active site volume is not significantly different (Table 1) which is also validated by the results of the analysis of the RMSF value showed that there was no change in the RMSF value of WT and mutants in the amino acid residues located at positions 190320 (area in the square line) (Figure 2a). The interaction of the ligand and active site protein in WT (Figure 4) used as example of docking illustration.
NE
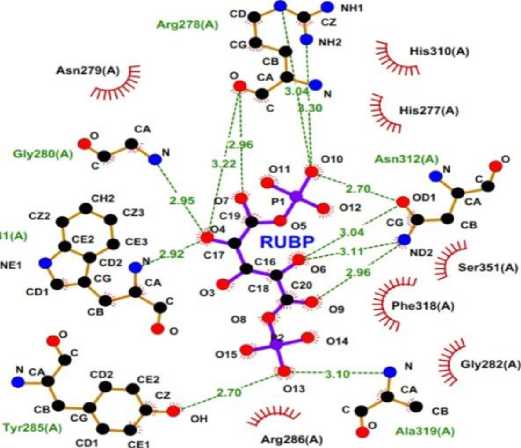
Figure 4. RUBP Ligand Interactions on The Active Site of RLP CS. Visualization Using LigPIot
CONCLUSION
The aspartic acid (asp) mutation at position 185 which is responsible for the formation of saltbridge (SB) with alanine (ala) and glutamic acid (glu) changes the structure of the RLP CS globally as indicated by a change in Rg value of mutant. Interestingly, although the global structure of the mutation results changes, the volume of the active region tends to remain the same and the interaction of the ligand-amino acid residues at the active site does not change as indicated by the binding energy values of the RLP WT and RMSF values of the MDS results of the two mutants which are similar. Aspartic acid mutation with alanine causes RLP CS to lose SB at position 185 which causes decreased stability (rising RMSD) and tends to fold more (low SASA value), this is consistent with the theory that SB is important for maintaining protein structure/conformation and stability.
ABBREVIATION
|
CS |
: Chromohalobacter salexigen BKL 5 |
|
MD |
: Molecular Docking |
|
MDS |
: Molecular Dynamic Simulation |
|
RLP |
: Rubisco Like Protein |
|
RMSD |
: Root Mean Square Deviation |
|
RMSF |
: Root Mean Square of Fluctuation |
|
Rg |
: Radius of Gyration |
RUBP : Ribulose Biposphate
SASA : Solvent Accessibilty Surface Area
SB : Saltbridge
WT : Wild Type
ACKNOLEDGEMENT
We thank the Institute Technology of Sumatera for awarding the scholarship and Hibah Bersaing Pasca Sarjana UGM for funding this study.
REFERENCE
Abraham, M.J., Murtola, T., Schulz, R., Páll, S., Smith, J.C., Hess, B., Lindahl, E. 2015. GROMACS: High Performance
Molecular Simulations Through MultiLevel Parallelism From Laptops to Supercomputers. SoftwareX. 1(2): 19– 25
Albeck, S., Unger, R., Schreiber, G. 2000. Evaluation of Direct and Cooperative Contributions Towards The Strength of Buried Hydrogen Bonds and Salt Bridges. Journal of Molecular Biology. 298(3): 503–520
Al-Karmalawy, A.A., Dahab, M.A., Metwaly, A.M., Elhady, S.S., Elkaeed, E.B., Eissa, I.H., Darwish, K.M. 2021. Molecular Docking and Dynamics Simulation Revealed the Potential
Inhibitory Activity of ACEIs Against SARS-CoV-2 Targeting the hACE2 Receptor. Frontiers in Chemistry. 9
Bosshard, H. R., Marti, D. N., Jelesarov, I. 2004. Protein Stabilization by Salt Bridges: Concepts, Experimental Approaches and Clarification of Some Misunderstandings. Journal of Molecular Recognition. 17(1): 1–16
Guex, N., Peitsch, M.C. 1997. SWISS-MODEL and the Swiss-Pdb Viewer: An
Environment for Comparative Protein Modeling. Electrophoresis. 18(15):
2714–2723
Hollingsworth, S.A., Dror, R.O. 2018. Molecular Dynamics Simulation for All. Neuron. 99(6): 1129–1143
Karplus, M., McCammon, J.A. 2002. Molecular Dynamics Simulations of
Biomolecules. Nature Structural Biology. 9(9): 646–652
Kumar, S., Nussinov, R. 2002. Close-Range Electrostatic Interactions in Proteins. ChemBioChem. 3(7): 604-610
Meng, X.Y., Zhang, H.X., Mezei, M., Cui, M. 2011. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Current Computer Aided-Drug Design. 7(2): 146–157
Messaoudi, A., Belguith, H., Ben Hamida, J. 2013. Homology Modeling and Virtual Screening Approaches to Identify Potent Inhibitors of VEB-1 β-Lactamase. Theoretical Biology and Medical Modelling. 10(1)
Miyazono, K., Tanokura, M. 2022. New Era in Structural Biology with The Alphafold Program. Translational and Regulatory Sciences. 4(2): 48–52
Morris, G.M., Huey, R., Lindstrom, W., Sanner, M.F., Belew, R. K., Goodsell, D. S., Olson, A.J. 2009. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. Journal of Computational Chemistry. 30(16): 2785–2791
Musafia, B., Buchner, V., Arad, D. 1995. Complex Salt Bridges in Proteins: Statistical Analysis of Structure and Function. Journal of Molecular Biology. 254(4): 761–770
Oostenbrink, C., Villa, A., Mark, A.E., Van Gunsteren, W.F. 2004. A Biomolecular
Force Field Based on The Free Enthalpy of Hydration and Solvation: The GROMOS Force-Field Parameter Sets 53A5 and 53A6. Journal of Computational Chemistry. 25(13): 1656–1676
Pettersen, E.F., Goddard, T.D., Huang, C.C., Couch, G. S., Greenblatt, D.M., Meng, E.C., Ferrin, T.E. 2004. UCSF Chimera: A Visualization System for Exploratory Research and Analysis. Journal of Computational Chemistry. 25(13):
1605–1612
Rácz, A., Mihalovits, L.M., Bajusz, D., Héberger, K., & Miranda-Quintana,
R.A. 2022. Molecular Dynamics Simulations and Diversity Selection by Extended Continuous Similarity Indices. Journal of Chemical Information and Modeling. 62(14):
3415–3425
Shukla, R., Shukla, H., Tripathi, T. 2018. Activity loss by H46A Mutation in Mycobacterium Tuberculosis Isocitrate Lyase is Due To Decrease in Structural Plasticity and Collective Motions of The Active Site. Tuberculosis.
108:143–150
Takano, K., Ogasahara, K., Kaneda, H., Yamagata, Y., Fujii, S., Kanaya, E., Kikuchi, M., Oobatake, M., Yutani, K. 1995. Contribution of Hydrophobic Residues to the Stability of Human Lysozyme: Calorimetric Studies and X-ray Structural Analysis of the Five Isoleucine to Valine Mutants. Journal of Molecular Biology. 254(1): 62–76
Takano, K., Tsuchimori, K., Yamagata, Y., Yutani, K. 2000. Contribution of Salt Bridges Near The Surface of A Protein to The Conformational Stability,. Biochemistry. 39(40): 12375–12381
Tian, W., Chen, C., Lei, X., Zhao, J., Liang, J. 2018. CASTp 3.0: Computed Atlas of Surface Topography of Proteins. Nucleic Acids Research. 46(W1):
W363–W367
Walker, K. D., Causgrove, T. P. 2009. Contribution of Arginine-Glutamic Acid Salt Bridges to Helix Stability. Journal of Molecular Modeling. 15(10): 1213–1219
7
Discussion and feedback