UTILIZATION OF SDT-RT-PCR FOR PLANT VIRUS DETECTION
on
J. Agric. Sci. and Biotechnol.
ISSN: 23020-113
Vol. 1, No. 1, Juli 2012
UTILIZATION OF SDT-RT-PCR FOR PLANT VIRUS DETECTION
I Gede Rai Maya Temaja*), Ni Made Puspawati, dan Ni Nyoman Ari Mayadewi
Faculty of Agriculture, Udayana University
*)
Corresponding author at: Jl. PB. Sudirman Denpasar Telp. (0361) 26532 E-mail address: tderai@yahoo.com
Abstrak
Metode simple-direct-tube (SDT) dikembangkan untuk preparasi RNA virus atau viroid, yang selanjutnya dipakai untuk sintesis cDNA. SDT tidak memerlukan penyaringan atau sentrifugasi seperi pada ekstraksi RNA total menggunakan KIT. Metode ini juga tidak menggunakan antiserum dalam preparasi RNA virus. Di samping itu metode SDT dapat dikerjakan di laboratorium dengan temperatur ruang dan selesai dikerjakan lebih kurang dalam 25 menit. Penerapan metode ini dilanjutkan dengan reverse transcription-polymerase chain reaction (RT-PCR), dapat mendeteksi keberadaan tomato chlorosis virus (ToCV), tomato infectious chlorosis virus (TICV), chrysanthemum virus B (CVB), Potyvirus and chrysanthemum stunt viroid (CSVd). Hal ini menunjukkan bahwa metode SDT dapat dipakai untuk mendeteksi virus atau viroid.
Key world: Tomato chlorosis virus (ToCV), Tomato infectious chlorosis virus (TICV), Chrysanthemum virus B (CVB), Potyvirus, Chrysanthemum stunt viroid (CSVd), metode simple direct tube, deteksi virus, RT-PCR
The reverse transcription-polymerase chain reaction (RT-PCR) and polymerase chain reaction (PCR) are powerful tools for highly sensitive detection of plant viruses with RNA and DNA genomes (Fenby et al., 1995; Singh, 1998). The necessary nucleotide sequences for the design of PCR primers are readily available for many plant viruses but nucleic acid extraction from plant tissues is a laborious and time-consuming step in RT-PCR procedures.
There have been many reports of simple and rapid techniques to detect plant viruses using RT PCR. Among them, direct binding (DB)-RT-PCR (Rowhani et a1.,1995), tube capture (TC)-RT-PCR (James, 1999) and simple-direct-tube (SDT)-RT-PCR (Suehiro et al. 2005) are easy and useful protocols to detect plant viruses without the use of phenol-chloroform extraction and antibodies, and are based on binding virus particles to the test tubes without any initial treatment. Because of DB- and TC-RT-PCR methods require several hours to obtain virus RNAs, attempts were made to improve
these techniques so as to simplify and shorten the time required for RNA extraction. Suehiro et al. (2005) has been developed a protocol (named the simple-direct-tube (SDT) method) that minimizes incubation times and omits centrifuging steps to obtain viral RNA from crude sap.
In order to apply and utilize this method for the detection of various plant viruses and viroid, representative important viruses, tomato chlorosis virus (ToCV; genus Crinivirus), tomato infectious chlorosis virus (TICV; genus Crinivirus), chrysanthemum virus B (CVB; genus Carlavirus), Potyvirus and chrysanthemum stunt viroid (CSVd) were examined as well.
Four plant viruses and one plant viroid, ToCV, TICV, CVB, Potyvirus and CSVd, collected in West Java Province, Indonesia were used in this study. ToCV and TICV were maintained in Lycopersicon esculentum L. Potyvirus was isolated from Pogostemon cablin. CVB and CSVd maintained in Dendranthema grandiflorum Kitam.
Virus-infected leaves were ground 1:1 (w/v) in the phosphate-buffered saline (PBS) solution containing 0.05% Tween-20 (PBST) (8 g sodium chloride, 1.15 g sodium phosphate dibasic, 0.2 g potassium phosphate monobasic, 0.2 g potassium chloride and 0.5 g Tween-20 dissolve in distilled water to 1000 ml, adjust pH to 7.4). The resulting crude sap (50 µl) was carefully placed into a PCR tube (0.5 ml, polypropylene) using a truncated tip to avoid trapping any air bubbles. After incubation at room temperature for 15 min, the sap was removed with a truncated tip. The tube was then washed twice with 50 µl of PBST to remove residual tissue. Thirty microlitres of diethylpyrocarbonate-treated water (DEPC-water) containing 15 units (U) of RNase inhibitor (Takara) was added to the tube, which was then immediately incubated at 95 °C for 1 min (denaturation), and cooled on ice for 1 min. The resulting solution was then used for RT-PCR. Crude sap from ToCV, Potyvirus and CSVd-infected leaves was used to evaluate the effect of different conditions on the standard procedure. The conditions examined was dilution of crude sap 101-, 102-, 103-, 104 - or 105fold in PBST.
cDNA was generated from virus/viroid RNA by reverse transcription (RT)
reaction. The reaction mixture consisting of a total of 10 µl (1 µl RT buffer 10x, 0.35 µl DTT 50mM; 2 µl dNTP 10 mM; 0.75 µl oligo dT 10 µM; 0.35 µl MMuLV Reverse transcriptase; 0.35 µl RNAse Inhibitor ; 2 µl virus/viroid RNA) was incubated at 42 °C for 1 h. One microlitre of the synthesized cDNA was added to 24 µl of the reaction mixture containing 2.5 µl of 10 x PCR buffer, 2.5 µl of 10 x sucrose cresol, 0.5 µl dNTP 10 mM, each of the 1 µl forward and reverse primers 10 mM, 1 µl of Taq DNA polymerase (1 U/µl) and 15.5 µl of sterile distilled water. Sequences of the primers and PCR cycling conditions used in this study are listed in Table l .
Table 1. Oligonucleotide primers for used in this study
|
Target |
Primer Sequence (5’-3’) |
Product size (bp) |
PCR cycling conditions | |
|
ToCV |
ToCV F: GTGTCAGGCCATTGTAAACCAAG ToCV R: ACAAAGCGTTTCTTTTCATAAGCAGG |
405 |
94°C, 4 min 94°C, 1 min 62°C, 1 min 72°C, 1 min 72°C, 10 min |
- 30 cycles |
|
TICV |
TiCV F: AATCGGTAGTGACACGAGTAGCATC TiCV R: CTTCAAACATCCTCCATCTGCC |
332 |
94°C, 4 min 94°C, 1 min 62°C, 1 min 72°C, 1 min 72°C, 10 min |
30 cycles |
|
Potyvirus |
PotyF: ATG GTH TGG TGY ATH GAR AAY GG PotyR: TGC TGC KGC YTT CAT YTG |
327 |
94°C, 2 min 94°C, 30 s 50°C, 1 min 72°C, 1 min 72°C, 10 min |
35 cycles |
|
CVB |
CVB5: CAAAGAGGTGATCATCCGTCTAG CVB3: CTCGGTTACTTTATCGCACCTAG |
739 |
94°C, 4 min 94 °C, 30 s 45 °C,1 min 72 °C,1 min 72°C, 10 min |
35 cycles |
|
CSVd |
CSVd-F: CAACTGAAGCTTCAACGCCTT CSVd-R: AGGATTACTCCTGTCTCGCA |
250 |
94°C, 4 min 96 °C, 30 s 61 °C, 45 s 72 °C, 1 min 72°C, 5 min |
- 35 cycles |
The SDT RT-PCR method is a very convenient technique to obtain template viral RNA for RT-PCR. ToCV, TICV, CVB, Potyvirus and CSVd were detected by the SDT-RT-PCR method, suggesting that particles of many different viruses or viroid can be directly immobilized onto polypropylene tubes within a few minutes (Fig. 1,2). The time required is less than 25 min before cDNA synthesis can be performed even when the number of samples is increased. This incubation period can be used to prepare the reverse transcription mixture.
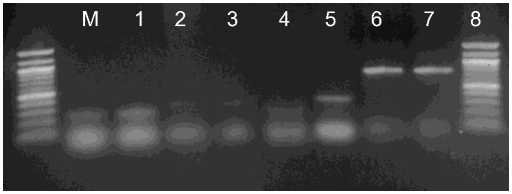
Fig. 1. Detection of CSVd (lanes 1-2), Potyvirus (lanes 3-4), TICV (lane 5), ToCV (lane 6) and CVB (lanes 7-8) by SDT-RT-PCR. Lane M: 100 bp DNA ladder (Fermentas)
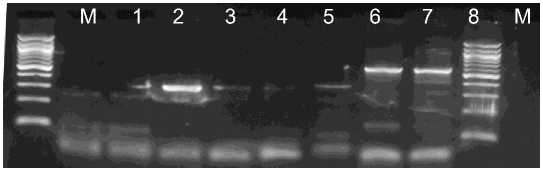
Fig. 2. Detection of CSVd (lanes 1-2), Potyvirus (lanes 3-4), ToCV (lanes 5-6) and CVB (lanes 7-8) by SDT-RT-PCR. Lane M: 100 bp DNA ladder (Fermentas)
The sensitivity of the SDT-RT-PCR method were stidied using serial dilutions of crude sap containing virus paticles. Agarose gel electrophoresis of the PCR product indicated that by the SDT-RT-PCR method were amplified when sap was diluted up to 103-, 104- and 105-fold for CSVd, Potyvirus and ToCV, respectively (Pig. 3,4). The DB- and TC-RT-PCR methods using direct immobilization described previously by Rowhani et al. (1995) and James (1999) have higher sensitivities than the SDT-RT-PCR method (Suehiro et al. 2005). However, much more time is required to obtain results with these methods. The objective of this study was to facilitate virus detection by obtaining template viral RNAs for RT-PCR by the shortest and simplest procedure. The SDT
method proved to be one of the fastest and most reliable protocols for detection of plant RNA viruses by RT-PCR.
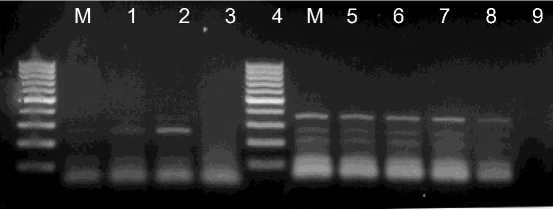
Fig. 3. Evaluation of dilution of crude sap prepared from infected leaves. Lanes 14; the crude sap prepared from CSVd infected leaves diluted 101-, 102-, 103-or 104-fold, respectively. Lanes 5-9; the crude sap prepared from ToCV infected leaves diluted 101-, 102-, 103-, 104- or 105-fold, respectively. Lane M: 100 bp DNA ladder (Fermentas)
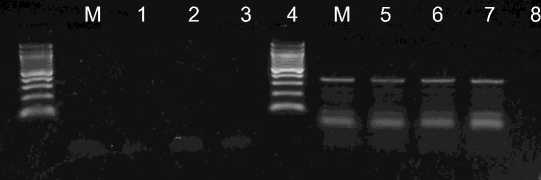
Fig. 4. Evaluation of dilution of crude sap prepared from Potyvirus infected leaves. Crude sap diluted 101-, 102-, 103- or 104-fold (lanes 5-8). Lane M: 100 bp DNA ladder (Fermentas)
The critical points for accurate detection by SDT-RT-PCR are washing the tube after crude sap incubation, to eliminate residual plant material completely; and timing the addition of DEPC-treated water and the denaturing period of the tube containing the immobilized virus particles (Suehiro et al. 2005). Plant species contain polyphenolic compounds or polysaccharides that affect the extraction of intact nucleic acids. It was possible to eliminate this problem by picking up visible material in the tube with a truncated tip (Thomson and Dietzgen, 1995; Suehiro et al. 2005). Depending on the circumstances, two or more washes may be required until the debris disappears. After washing, PBST should be completely removed using normal tips. It is important to avoid degradation of the viral RNA in the tube before cDNA synthesis, so DEPC-water should be added after preparing the RT-reaction mixture and adjusting the temperature of heat block.
Adsorption of virus particles to the tubes is thought to be caused by non-
specific attachment, causing SDT-RT-PCR is very useful tool for the detection of viruses especially when PCR primers for the target virus RNA are available but antisera against the virus particles are not. For example, the method might be available to detect viruses with universal primers for cucumoviruses (Choi et al., 1999) and potyviruses (Chen et al., 2001). It was demonstrated by Suehiro et al. (2005) that a large fragment at the 5' region of the TuMV genome was amplified by SDT-RT-PCR, suggesting that it may be possible to synthesize the full-length cDNA of TuMV. This research suggest that the rapidity and simplicity of the SDT method could make it useful in the detection of virus pathogens.
Acknowledgement
This study was supported by Laboratory of Virology, Department of Plant Protection, Faculty of Agriculture, Bogor University of Agriculture. I thank Ms. Tuti Susanti Legiastuti for their excellent technical assistance.
References
Fenby, N.S., Scott, N.W., Slater, A., Elliott, M.C., 1995. PCR and nonisotopic labeling techniques for plant virus detection. Cell. Mol. Biol. 41, 639-652.
Chen, J., Chen, J., Adams, M.J., 2001. A universal PCR primer to detect members of the Potyviridae and its use to examine the taxonomic status of several members of the family. Arch. Virol. 146, 757-766.
Choi, S.K., Choi, J.K., Park, W.M., Ryu, K.H., 1999. RT-PCR detection and identification of three species of cucumoviruses with a genus-specific single pair of primers. J. Virol. Methods 83, 67-73.
James, D., 1999. A simple and reliable protocol for the detection of apple stem grooving virus by RT-PCR and in a multiplex PCR assay. J. Virol. Methods 83, 1-9.
Kamenova, I., and Adkins, S. 2004. Comparison of detection methods for a novel tobamovirus isolated from Florida hibiscus. Plant Dis. 88:34-40.
Rowhani, A., Maningas, M.A., Lile, L.S., Daubert. S.D., Golino, D.A., 1995. Development of a detection system for viruses of woody plants based on PCR analysis of immobilized virions. Phytopathology 85, 347-352.
Singh, R.P., 1998. Reverse-transcription polymerase chain reaction for the
detection of viruses from plants and aphids. J. Virol. Methods 74, 125-138.
Suehiro, N., Matsuda, K., Okuda, S., Natsuaki, T., 2005. A simplified method for obtaining plant viral RNA for RT-PCR. J. Virol. Methods 125, 67-73.
Thomson, D., Dietzgen, R.G., 1995. Detection of DNA and RNA plant viruses by PCR and RT-PCR using a rapid virus release protocol without tissue homogenization. J. Virol. Methods 54, 85-95.
http://ojs.unud.ac.id/index.php/JASB
29
Discussion and feedback