DNA Probe Design for Detection Mutation at Codon 315 In katG Gene of Mycobacterium Tuberculosis to Real-Time Polymerase Chain Reaction
on
Journal of Health Sciences and Medicine, Vol. 1 No. 2, September 2017
31
DNA Probe Design for Detection Mutation at Codon 315 In katG Gene of Mycobacterium Tuberculosis to Real-Time Polymerase Chain Reaction
I Gusti A. A. Santhi Rahmaryani1, Ni Kadek Ariani1, Dyah Subadrika Warma Dewi1, Ni Komang Sasi Ani1, Ade Ari Sundari1, Kadek Widya Yuli Hartati1, and Sagung Chandra Yowani2
-
1Department of Pharmacy, Faculty of Mathematics and Natural Science, Udayana University, Bali, Indonesia Email: agayusanthi@gmail.com
-
2 MDR-TB & XDR-TB Research Group
Faculty of Mathematics and Natural Science, Udayana University, Bali, Indonesia Email : cyowani@gmail.com
Abstract:
High-level resistance to isoniazid as a first-line tuberculosis drugs can be caused by mutations in codon 315 katG Mycobacterium tuberculosis. Mutation at codon 315 is the most frequent mutation with the highest amino acid variation, compared to other codons in the Mycobacterium tuberculosis katG gene. Therefore, a specific probe is required for rapid and proper detection of mutations at codon 315. In this study, the design of a nucleotide sequence probe with TaqMan labeling was performed using Clone Manager Suite 6 software. The mutant probe obtained was analysed in two stages. The initial analysis is based on the length of the probe (22-30 bases), Tm (70ºC), %GC (35-65%), not in hairpin form, dimer (< 5 bases), runs and repeat (≤ 4 for base A, T, C, and < 3 for base G). Furthermore the final analysis was carried out with no G base in 2 bases at the end of the 5’ probe and the amount of base C ≥ G.
The study resulted in 260 probe mutants. After the initial analysis, 11 mutant probes were obtained to recognize mutations in the codon of 315 katG Mycobacterioum tuberculosis genes. The probe consists of 2 probes for the S315T mutation, 6 probes for S315N mutation, and 3 probes for S315V mutation. The criteria of the 11 mutant probes are 22-23 bases long, Tm 70ºC, % GC 56-63%, 4 dimer, 2 runs, and does not have repeats and does not form hairpin at a temperature of 56ºC. Based on the final analysis, 3 mutant probes were obtained fulfilling the TaqMan probe labeling criteria, namely K315MT1 for specific detection of mutation S→T and K315MN5, then K315MN23 for specific detection of mutation S→N.
The conclusion of this study shows that the best mutant nucleotide sequence probes for the detection of mutations at codon of 315 KatG Mycobacterium tuberculosis genes are 5’-FAM-CC ACC GGC ATC GAG GTC GTA TG-TAMRA-3’; 5’FAM-ATC ACC AAC GGC ATC GAG GTC G-TAMRA-3’; dan 5’FAM-C ACC AAC GGC ATC GAG GTC GTA T-TAMRA-3’. The design of the mutant probe according to the TaqMan probe criterion for real-time PCR was obtained by 3 probes from the 11 selected mutant probes in the initial analysis.
Keywords: Tuberculosis, gene katG mutation, in silico, TaqMan probe, real-time PCR.
-
I. INTRODUCTION
According to World Health Organization survey data in 2014, Tuberculosis (TB) is the second leading cause of death in the world after Acquired Immuno Deficiency Syndrome (1). TB control is increasingly difficult with the onset of Multi-drug Resistant Tuberculosis (MDR-TB), which is defined as tuberculosis caused by Mycobacterium tuberculosis resistance to at least two first-line antituberculosis drugs (OAT), such as isoniazid and rifampicin (2). Resistance to isoniazid is found in high frequency. According to research by Lisdawati et al., (2015) the percentage of resistance to isoniazid in Indonesia is 28.6% (3).
Resistance to isoniazid can be caused by mutations in some Mycobacterium tuberculosis genes, such as katG genes, inhA (4), ahpC, kasA and ndh (5). Mutations in the katG and inhA genes can be used as predictive markers to predict resistance to isoniazid. KatG gene is the most commonly mutated gene with the percentage of 78% (6). Therefore, the katG gene is the focus of this study.
Mutations in the katG gene resulted in amino acid changes of the catalase-peroxidase enzyme which decreased the activity of the catalase-peroxidase enzyme in converting isoniazid into its active form, so that isoniazid resistance (7,8) occured. The codon 315 in the most commonly mutated katG genes and amino acids formed by mutations in
codon 315 has the greatest variation among the codon of other katG genes.
Two studies conducted on MDR-TB isolates in Bali with an amplified region of 2437-3160 pb showed that, the amino acid variation of codon 315 was due to mutations of S315T and S315V (9.8). Other mutations in this codon that cause variations in amino acids, was shown in the study of Bostanabad et al. (2008) to isolates in Belarus which include S315R, S315N, and S315G mutations. Mutation S315T has the highest prevalence. The research of Rintiswati et al. (2011) showed that the S315T mutation was found in 27.05% of sputum samples obtained from several laboratories in Indonesia. The other studies also showed that the frequency S315T in clinical isolates that were resistant to isoniazid was very high at 46-85% (10,11,12).
Detection of Mycobacterium tuberculosis which is resistant to isoniazid can be done by molecular method; one of them is Real-Time Polymerase Chain Reaction (real-time PCR). Real-Time PCR is a combination of PCR chemical methods with fluorescence molecules to monitor the amplification products in each PCR cycle. The real-time PCR detection system is divided into 2 fluorescence molecules that are interconnected in a DNA double strand, and oligonucleotides is labeled fluorescence compound called DNA probe (13). The real-time PCR method with DNA probe is capable of providing sensitivity and selectivity equivalent to conventional PCR analysis methods combined with southern blot (14). In addition, the fluorescence signal generated by the primer dimer would not be detected with the use of DNA probes. Therefore, the DNA probe is more specific than the intercalated fluorescence molecule in the DNA double strand (15).
There are several kinds of DNA probe detection systems used for real-time PCRs, one of which is TaqMan probe (13). TaqMan probe is a type of hydrolysis probe since it involves the activity of exonuclease from Taq DNA polymerase. The sequence of the nucleotide probe with two kinds of hybridized fluorescence labels will be degraded by the exonuclease activity resulting in a fluorescence signal (16). The advantage of TaqMan probe is safer than radioactive probe (17), the detection result can be known quickly (18) and it is specific. TaqMan probe has a good specificity because the sequence of the nucleotide probe will be hybridized with the target sequence on the amplification product (19).
The design of the DNA probe is a critical stage for all detection stages based on the hybridization process to ensure the stickiness specificity and detection efficiency. The good result of detection with DNA probe depends on the design criteria of DNA probe used. Some of the required criteria are probe length, guanine and cytosine base composition (G-C), mutation position, hairpin formation, and availability of a single base or dinucleotide over a long range exceeding 4 (16). The sequence of the DNA probe must also be specific to the target DNA sequence. Therefore, DNA probe design is required to avoid cross hybridization in the sequence that has many equations (20).
The use of software in the design of nucleotide probes sequence can dynamically optimize the design results compared to the manual design. The design of nucleotide probes sequence with methods based on trial and error in the laboratory, the availability of physical needs such as tools, reagents and the number of target cloning or nontarget microbial groups will result in optimization of a nucleotide probes sequence getting more complicated. DNA probe designs made with software can be used as a reference to make experimental DNA probes in the laboratory. In addition, DNA probe design using the software will be able to minimize the time, and maximize the probability of successful proliferation of DNA probe with target DNA sequence as an attempt to detect mutations in KatG Mycobacterium tuberculosis gene (21,22).
The study is designed a DNA probe to detect mutations in codon 315 on katG Mycobacterium tuberculosis genes. The best DNA probes will be selected from several design results using the Clone Manager Suite 6 software and the design criteria are also matched against DNA probe criteria based on the literature. The selected DNA probes will be applied for rapid detection of resistance to isoniazid using real-time PCR method.
-
II. MATERIAL AND METHODS
MATERIAL
The probe design requires nucleotide sequence of katG Mycobacterium tuberculosis H37Rv (wild-type) genes from database URL://www.ncbi.nlm.nih.gov (accession number: X68081.1). A primer pair for probe analysis consists of a forward primer with a sequence of 5’-GAAGTACGGCAAGAAGCTCTC-3’ and a reverse primer a sequence of 5’-CGATCTATGAGCGGATCACG-3’. The primer is the best design in silico and has been experimentally optimized for the detection of mutations in the Mycobacterium tuberculosis katG gene. The experimental optimization results showed that at an annealing temperature of 56ºC, the primers can constantly produce thicker DNA bands and do not form dimers (Deniariasih, 2013; Suryadi, 2013). In this DNA probe design study, we used long data and Tm primer in silico as reference to determine the design criteria of DNA probe.
TOOLS
The equipment to complete the research is a set of laptops with Windows 10 32 bit specifications, modems, and Software Clone Manager Suite 6 (University of Groningen) for designing and analyzing DNA probes.
STEPS
Sequence of Nucleotide Targets for Designing Probe DNA Probes
The design of the probe DNA uses the whole sequence of the genotype katG gene composed of 4810 pb with the starting codon at 1979 pb. DNA probes will be designed inside the amplification area at position 2437-3160 pb katG Mycobacterium tuberculosis genes. In Figure 1, it
can be seen that the amplification region is a nucleotide sequence in the light blue line, numbered based on the total number of Mycobacterium tuberculosis katG genes. The amplification areas are limited by a pair of primers marked in green and include the position of mutations in the Mycobacterium tuberculosis katG gene based on Deniariasih's (2013) and Suryadi (2013) studies. One of the most commonly coded codons is the 315 codon (AGC). Therefore, codon 315 will serve as a specific design target of the mutant probe. The position of codon 315 is indicated by yellow in Figure 1.

Figure 1. Nucleotide sequence of Mycobacterium tuberculosis katG gene targeted for DNA probe design (8).
Mutations in codon 315 are found in several variations, namely S315T, S315N, S315R, S315G, and S315V. This mutation results in a nucleotide change of the 315 codon. Therefore, the design of the DNA sequence of the mutant probe refers to the nucleotide change of the codon 315. The nucleotide changes to codon 315 and its positions are listed in Table 1.
Table 1. Changes of Nukleotida Kodon 315 Mycobacterium tuberculosis katG Genes for DNA Probe Design Mutants
|
No. |
Probe Name |
Codon 315 |
Mutation |
Nucleotides |
Position from codon start | |
|
wild-iype |
Jstoaa | |||||
|
1. |
K315MT |
AGC |
S315T |
G |
C |
944 ⅛ |
|
2. |
K315MN |
S315N |
G |
A |
944 ⅛ | |
|
3. |
K315MR |
S315R |
C |
G |
94: ⅛ | |
|
4. |
K315MG |
S315G |
A |
G |
943 ε⅛ | |
|
5. |
K315MV |
S315V |
AG |
GT |
943, 944 Eh | |
Information: Probe name, e.g. K315MT: K = denotes M. tuberculosis katG genes; 315 = denotes the targen codon; M = denotes the type of probe design is a mutant probe; T = amino acids formed by mutations at codon 315
Designing DNA Probe
The design of DNA probes was conducted in silico by software Clone Manager Suite 6. The first step is to download the nucleotide sequence of Mycobacterium tuberculosis H37Rv (wild-type) with accession number: X68081.1 in FASTA data format and saved in the notepad as a template. Next opened the Clone Manager Suite 6 program and clicked the primary menu, select the design, and then entered the data sequence nucleotide katG wildtype gene that has been stored in notepad and clicked OK. In the primary type, the probe is selected, then the length of the probe is varied from 22-30 bases in the length section and set the target range range including the mutation at codon
-
315, then clicked OK. Furthermore, some DNA probe designs will appear, then clicked primer report to display the nucleotide sequence of the probe. Nucleotide sequences that include the position of the mutations were screened. At this stage, some wild-type probe DNA designs will be produced. One base on the wild-type probe was subsequently replaced by mutation at codon 315 (Table 1) in order to obtain sequences of mutant probe nucleotide for the detection of mutations in the 315 codon Mycobacterium tuberculosis katG genes.
-
III. RESULTS AND DISCUSSION
The Sequences of Nucleotide Target for Designing Probe DNA Probes
The codon 315 was the target of the nucleotide sequence design of the mutant probes in this study. Mutations in the codon of 315 Mycobacterium tuberculosis katG genes are one of the mutations that cause resistance to isoniazid. The resulting resistance level is high enough to cause the MIC of isoniazid to > 5 µg/ml (23). The prevalence and variation of amino acids from this codon mutation is also the highest compared to other codons in the Mycobacterium tuberculosis katG gene, such as codons 309, 316, and 357 (11). The prevalence of mutations at codon 315 reached 74% (12). Meanwhile, amino acid variations are formed due to several mutations at codon 315 with different prevalence, e.g. S315T 36-85%, S315N 1,9-5,7%, S315R 0,9-2,4%, S315G 1,1-2,4%, and S315V 1% (10, 11, 24, 9).
The nucleotide sequence used to design the mutant probe is the overall wild-type nucleotide sequence of the Mycobacterium tuberculosis katG genes. This wild-type nucleotide sequence is obtained online from genbank code X68081.1. Genbank is a public database managed by the National Center for Biotechnology Information (NCBI). A number of online services to identify, align, and compare sequences are also provided by NCBI (25). In the previous study, the overall sequence of nucleotides of this katG gene has been used in the primary design for the detection of mutations in TB isolates in Bali with PCR. Based on these studies, two isolates were found to have mutations at codon 315 (8, 9).
Results of DNA Probe Design
The probe is a complementary nucleotide sequence with a nucleotide sequence on the target such as a primer. However, the probe does not limit the PCR amplification region and can not be prolonged as it has been phosphorylated at the 3 'end. In addition, the probe can also be used for detection because it is labeled with a pair of dyes in the form of a reporter and quencher at both ends and will be hydrolyzed when the elongation process proceeds (26).
The design of the mutant probe DNA is designed using Clone Manager Suite 6 Software. The probes can be generated with initial criteria, ie GC and Tm values, dimer, runs, repeats, and hairpin. Initial criteria software is not absolute, so it can be tailored to the needs. The design of
nucleotide sequence probe using this software has not been done. In previous study, this software has been used to design a pair of primers for the detection of mutations in the Mycobacterium tuberculosis katG gene (9, 8). Therefore, in this study, it is proved that Clone Manager Suite 6 Software can generate a TaqMan nucleotide probe sequence. This research is made easier because of the availability of this software along with the license code for software activation. Some other software for probe design such as Alleleid, Beacon Designer, Oligo7, and Probe Maker are not used in this study because the license code of the software is inaccessible to the researcher, so it can not be used completely.
The designed mutant probe is a TaqMan probe. This type of probe has a simple design and the fluorescence can be determined at the end of the extension phase (13). The TaqMan probe design is a linear oligonucleotide arrangement labeled by reporters at the 5 'end and quencher at the 3' end (17). TaqMan probe is specific so it can be used to detect a base change in the target nucleotide sequence (27). The fluorescence signal will increase in proportion to the increasing distance between the reporter and the quencher of the hybridized probe. The increase in the distance of the two labels occurs by probe hydrolysis by the 5 '→ 3' exonuclease activity of Taq polymerase enzyme during PCR (15).
The design of a mutant probe with Clone Manager Suite 6 software begins with the design of a wild-type probe. The resulting wild-type probe design results do not specifically include the desired target mutation area. Therefore, selection based on target area coverage was performed to obtain a wild-type probe design covering the range of mutation areas at 943, 944, and 945 pb position of the start codon (or position 2921, 2922, and 2923 pb of the whole katG gene). Screening of the mutation position against the wild-type probe design is also performed. The position of the mutation affects the hybridisation of the nucleotide sequence of the probe. The probe hybridization with mismacth in the center of the nucleotide sequence of the probe is more unstable than the probe hybridization with a mismatch near the probe tip 28. In the event of a mismatch between the probe and the target, the probe hybridization efficiency will decrease. In this condition, Taq polymerase enzyme will lengthen the probe target without degrading the probe, so the reporter fluorescence is undetectable (29, 30). The mismatch relationship with hybridization is shown in Figure 2. Detection of allele 1 (black mutation position) using a probe with a difference in one type of nucleotide (white mutation position in the center of the probe) resulted in a mismatch. The probe-target hybridization becomes unstable under mismatch conditions. Therefore, probe degradation cannot occur and FAM fluorescence signals (reporter) are not generated.
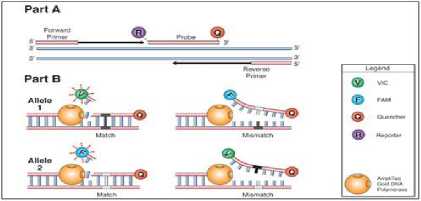
Figure 2. Effect of Mutation Position on Probe Hybridization
Caption:(Part A) In the DNA amplification process, primers and probes will hybridize with their complementary nucleotide sequence on the target DNA; (Part B) The presence of a mismatch causes the probe not to be degraded so that the fluorescence signal is not formed (Anonymous d, 2001)
The selected wild-type probe design is then converted to mutant probe design via nucleotide base replacement in the direct entry menu. Replacement is made to obtain a specific mutant probe design for nucleotide change at codon 315. Each mutant probe made is marked by name. For example for mutant probe 1 with mutation S315T is named K315MT1. K denotes the KatG Mycobacterium tuberculosis genes, 315 is the target codon, M denotes the mutant probe, T denotes the name of the amino acid due to mutation and the number 1 is the serial number of the mutant probe design. There are 260 probes mutant probes generated from the design process in silico. The design of the mutant probe consists of probes for several mutations at codon 315, such as the S315T, S315N, S315R, S315G, and S315V mutation.
Result of Mutable Probe Design Analysis Initial Analysis Result of Mutan Probe
The analysis process was conducted in two stages consisting of initial analysis and final analysis. Gradual analysis is intended to maximize the selection process of probe designs in silico. An initial analysis was performed to obtain a mutant probe design that meets the general criteria of the probe. Analyze process used direct entry menu. Designs that meet all general criteria of the probe will be displayed with a check mark (√) on the software, whereas if any of the criteria does not match, then the results of the analysis will be displayed with a cross (×). An example of the analysis results was shown with the software in Figure 3. Figure A shows the results of initial analysis of K315MN23 probe met probe criteria, whereas in figure B the results of K315MN30 probe analysis did not meet the probe Tm criteria. In addition to the results also displayed the sequence of nucleotides of each mutant probe.

Figure 3. Example display of early stage analysis results in software
Caption: (A) The nucleotide sequence of the K315MN23 probe meets the criteria of the initial probe analysis of the software; (B) The nucleotide sequence of the K315MN30 probe does not meet the Tm criteria of the software
Some of the criteria for initial analysis of the probe are based on the primary criterion used (15). The primary criteria used as a reference in this study are the length and value of primary Tm to determine the length and value of Tm probe. This is necessary to prevent non-specific attachment of primers or probes as they are in the same reaction during PCR (31). The initial criteria of the analysis included the length of the 22-30 base probe, the Tm 70ºC value, the GC content of 35-65%, the dimer less than 5 bases, the hairpin was not formed, and the number of runs and repeats was not more than 4 for bases other than guanine base. Special runs and guanine base repeats should be less than 3 (15, 16).
The results of the initial analysis of the mutant probe obtained were 11 mutant probes for mutation detection at codon 315. The criteria and sequence of mutant probes were shown in Table 2. All the mutant probes (Table 2) had variations in lengths from 22-23 bases. The overall design meets the length of TaqMan probe criteria. Generally TaqMan probes are designed with lengths that exceed the length of primary amplification. The maximum length of a good TaqMan probe is 30 bases (16). The use of probes of length exceeding the primer is intended to obtain a higher probe Tm than the primary Tm (27). This is intended to prevent inefficient degradation of the probe. One of the reasons is that the upstream primer undergoes an extension process before the probe hybridizes with target (32). The longer the probe size, the time required for annealing will be longer (33). The specificity of PCR can also be affected by probe length. Probes that are too long are less specific to distinguish mismatch. This can lead to false-positive results, especially when the difference between 2 or more probes used lies solely on single nucleotide (27). Conversely, the shorter the probe, the annealing time is shorter, but the attachment of the probe to the undesirable area (mispriming) will increase (34).
Table 2. Results of Initial Analysis of Mutant Probe For Codon 315
|
Probe ∖'amβ |
Nucleotide sequence of JVafee 5—»3’ |
DNA Probe Design Criteria ⅛ .⅛⅛⅛j? | ||||||
|
Ejg |
Tm |
% |
Dimei- |
Jiabpin |
raws |
> spear | ||
|
Probe design of specific Kiidation. S315T | ||||||||
|
K315MTI |
CCACOGGCATCGAGGrCGTATG |
22 |
70 |
63 |
4 |
- | ||
|
K315MT4 |
Gatcaccaccggcatcgaggtc |
ZZ |
,iθ |
63 |
4 |
- | ||
|
Frcbe design of specific mutation S315N | ||||||||
|
K315MN3 |
Cgatcaccaacggcatcgaggt |
S |
.l0 |
59 |
4 |
- |
Z | |
|
K315MN5 |
Atcaccaacggcatcgaggtcg |
22 |
,1'0 |
59 |
4 |
- |
Z | |
|
K315MN16 |
Ccaacggcatcgaggtcgtatgg |
23 |
70 |
60 |
4 |
- |
Z | |
|
K315MN17 |
CAACGGCATCGAGGrCGTATGGA |
23 |
70 |
56 |
4 |
- |
Z | |
|
K315MN23 |
Caccaacggcatcgaggtcgtat |
23 |
70 |
56 |
4 |
Z | ||
|
K315MN24 |
Accaacggcatcgaggtcgtatg |
23 |
70 |
56 |
4 |
- |
Z | |
|
J⅛S⅛S J»®*® ⅛h⅛i½ mutasιS315 V | ||||||||
|
K315MV1 |
Ccgtcggcatcgaggtcgtatg |
22 |
70 |
63 |
4 |
- |
2 | |
|
K315MV4 |
Gatcaccgtcggcatcgaggtc |
22 |
'.,0 |
63 |
4 |
Z | ||
|
K315MV8 |
ACCGrCGGCATCGAGGTCGTAT |
22 |
.0 |
59 |
4 | |||
Information: The probe name, for example K315MT1 shows: M. tuberculosis katG genes; 315 = target codon; M = type of probe is a mutant probe; T = amino acids formed by mutation of codon 315; number 1 = serial number of the probe design; Pjg = length; Tm = melting temperature (ºC); % GC = percentage of guanine and cytosine base content; underscore = mutation at codon 315
All mutant probe designs are then selected based on Tm values. Tm values can be used to predict the annealing temperature (Ta) of the primer or probe. In general, Ta can be determined as a temperature of 5ºC below the estimated Tm (35). A good Tm value for TaqMan probe is 5-10ºC higher than the primary Tm amplification value (31, 36). A higher probe probe will cause the probe to remain hybridized with the target DNA prior to the primary elongation process. The optimum Tm for the probe design to be effectively hybridized with the target DNA is ~ 8ºC over the primary Tm (16) or 70ºC (37,38). In this study, there were 85 mutant probes have a Tm of about 67-72 º C. The probe meets the range of Tm TaqMan probe criteria, which is Tm 5-10ºC above the primary. However, in order to obtain the best mutant probe, in this study re-selection based on the optimal Tm probe value in general (70ºC). The number of mutant probe designs meeting the optimal Tm criterion was 13 probes. After initial analysis, there were 11 mutant probes with optimal Tm that satisfied all of the probe's initial criteria (Table 2). The number of optimal mutant probe designs based on Tm for nucleotide changes at codon 315 varied. Changes in nucleotide composition occurred due to mutation variation at codon 315. The more GC content and the longer the probe, the Tm of the probe increased (39).
The third criterion assessed for obtaining good mutant probe design is the GC content. The requirement of a good GC content for the probe is 35-65% (15). The mutant probe obtained from the initial analysis (Table 2) has met the requirements of a good GC probe content. GC content outside the criterion range may cause instability of probe hybridization in the target area during the elongation process (26), and increase non-specific hybridization thereby decreasing the efficiency of PCR. The relationship between
Tm and GC content are seen in the design of the mutant probe for the S315G mutation. This type of probe has the highest Tm compared to other mutant probe designs though at the same length. Therefore, in this study no optimal mutant probe design for S315G mutation was obtained.
All subsequent designs were analyzed based on the general criteria of number of runs and repeats. The fewer the number of runs and repeats, the better the probe design is generated (40). The number of runs and repeats that can be accepted as a good probe is not more than 4 (16). Special base guanine, repetition sequentially as much as 3 or more must be avoided. 3 or more sequential guanine bases can fold the template into a tetraplex structure. This structure is very stable and cannot be recognized by the polymerase enzyme. Secondary structures are also formed when repeats are present. Repetition of the long dinucleotide can produce a complementary sequence to form a structure such as a hairpin (41). In addition, the number of runs and repeats that exceed the criteria will also increase the cost for detection (42).
The mutant probe design that meets the number criteria of runs and repeats is the mutant probes design for mutations S315T, S315N, and S315V. The maximum number of runs and repeats on each mutant probe design is 2. While the mutant probe design for the S315R mutation does not meet this criterion since it has 4 runs of the guanine base. Runs of the guanine base occurred in all mutant probe designs for the S315R mutation. A single base changed at the 315 codon from AGC to AGG and affected the base composition of this probe, resulting in 4 runs of the guanine base, as shown in Figure 4. In the figure, there were wildtype (WT) probe and K315MR8 mutant probe design (MT) with the position of mutation C → G (marked x) resulted in the addition of the number of G on the sequence of the mutant probe and formed the runs.
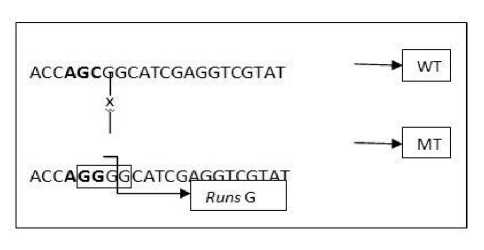
Figure 4 Runs on the K315MR8 mutant probe design
Caption: runs of 4 bases G on K315MR8 mutant probes are formed due to nucleotide changes C → G at codon 315. The formation of runs of 4 base Gs does not result in a nucleotide sequence of the K315MR probe that meets the initial stage analysis criteria for the TaqMan probe
Other criteria that also need to be considered to produce a good mutant probe are dimer and hairpin. The probe should not contain complementary bases so it can form a secondary structure, such as dimer and hairpin (17). Dimers are formed by intermolecular interactions between
two probes (43). The formation of dimers can occur in all mutant probes obtained. The type of dimer formed was a self dimer of 4 nucleotide bases. The dimer formation of one of the mutant probes, e.g. K315MN23 is shown in Figure 5. The dimer of the K315MN23 mutant probe is formed between the TCGA bases with its complementary base on the same probe (indicated by dashed lines in Fig. 5). Selected mutant probes based on early stage analysis (Table 2) have met the initial criteria of dimer count in Clone Manager Suite Software 6. Initial criteria for allowed dimers are less than 5 bases. However, it should be noted that dimer formation in the probe design can reduce hybridization efficiency resulting in decreased detection sensitivity (15).
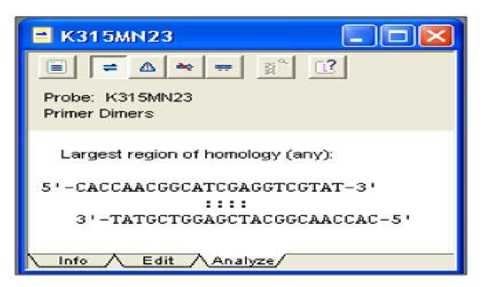
Figure 5 The result of dimer analysis on K315MN23 mutant probe design
Caption: All nucleotide sequences of the mutant probe form 4 dimers, including the K315MN23 probe and have met the criteria of good probe dimer quantities (<5)
Hybridisation efficiency can also be reduced if the probe design forms another secondary structure, such as a hairpin. The curved hairpin structure can inhibit TaqMan hybridization probe process at target (17). The formation of the hairpin from the nucleotide sequence of the probe is shown by showing the free structure and energy (ΔG) image at the specific temperature set in Clone Manager Suite 6. Hairpin is affected by the annealing temperature used. The increasing annealing temperature can melt and reduce the formation of the hairpin structure. This occurs in the design of the resulting nucleotide probe sequence. At annealing temperatures from 1 to 36ºC, the nucleotide sequence of the mutant probe (Table 2) forms hairpin with different free energies at different temperatures. For example, a hairpin formed from the nucleotide sequence of the K315MT1 probe at 10ºC (Figure 6A) and 20ºC (Figure 6B) is shown with a red circle in Figure 6. The ΔG value at 10ºC is -0.6 kcals, whereas at 20ºC is - 0.1 kcals. Although hairpin with negative ΔG must be avoided, but the resulting mutant probe design meets the maximum hairpin criteria ΔG because the free energy of acceptably stable hairpin is -3 kcals (40, 44). In addition, all mutant probe designs do not form hairpins at annealing temperatures that are adjusted according to an experimental primer annealing temperature of 56ºC. Therefore, all nucleotide sequences of mutant probes have met the criteria of the secondary structure of hairpin.
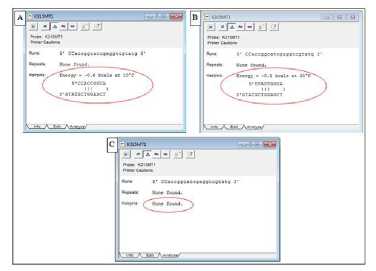
Figure 6. The hairpin structure depiction formed by the K315MT1 mutant probe with Clone Manager Suite 6
Caption: the red circle shows the hairpin structure and its free energy; (A) The structure of a hairpin K315MT1 at a temperature of 10ºC; (B) The structure of a hairpin K315MT1 at a temperature of 20ºC; (C) The hairpin structure is not formed at 56ºC, which is the primary optimization Ta based on Suryadi's experimental study (2013)
The Analysis on Labeling of Mutant Probes
Select mutant probe designs in the initial analysis (Table 2), then was analyzed the final stages. The final analysis criterion is based on the TaqMan probe labeling that there is no base G on the 2 bases at the 5 'end of the probe, and the amount of base C is more or equal to the base G on the nucleotide sequence of the probe. A final analysis is needed to find out the suitability of the probe design with TaqMan probe criteria and to produce a quality TaqMan probe. One of the factors that determine the TaqMan probe's quality is the fluorescence blackout efficiency of the probe label (26).
The efficiency of blackout of a good reporter label is influenced by several factors, such as the optimal distance between the two labels, as well as the number and position of the guanine base (16, 28, 33). Reporter blackouts can be performed optimally by quencher compounds located at a distance of 3-30 bases from the reporter. Quencher at a 2-base distance from the reporter will result in the Taq polymerase enzyme, which is not able to perform the exonuclease activity that plays a role in the degradation of the probe to produce a fluorescence signal (28). The basic structure G contains most atoms with electronegative properties that can contribute a free electron pair. This will result in the greatest electron-donor electron ability compared to other nucleotide bases. Therefore, the amount and position of base G are very necessary to be considered in order to obtain optimal signal detection efficiency (45).
The resulting mutant probe should not contain base G on the first and second nucleotides from the 5 'probe end (44). This criterion is important because the guanine base has a natural ability to extinguish fluorescence (quencher) from reporters such as FAM or other fluorescent compound derivatives. Based on the criteria of position G on the nucleotide sequence of the probe, 8 probes were found that met this criterion, of the 11 mutant probes in Table 2
analyzed. A probe that does not meet this criterion is 3 probes with position G as shown in Figure 7. The base G at the nucleotide sequence of the three probes is at the 1st position of the 5 'end for K315MT4 and K315MV4 probes, and at the 2nd position of the tip 5 'for K315MN3 probe.
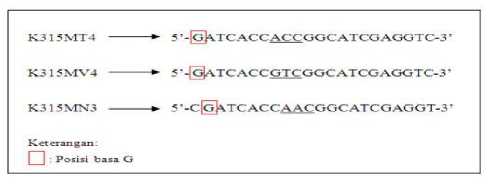
Figure 7. The base position of G on the K315MT4, K315MV4, and K315MN3 probes
Caption: These three mutant probes do not meet the final stages of analysis criteria, ie the minimum G position at the 3rd base from the 5 '
The second criterion for the final analysis of the probe is the amount of base C more or equal to the base G on the nucleotide sequence of the probe. In the 11 mutant probes analyzed in the final analysis, there were 5 mutant probes obtained such as K315MN16, K315MN17, K315MN24, K315MV1, and K315MV18 had fewer C than base G. Based on empirical observations, it was found that TaqMan probe with a higher number of G bases than base C is often resulting in decreasing normal fluorescence (40). This relates to the nature of a base G which can extinguish the reporter fluorescence, although the nucleotide sequence of the probe has been hydrolyzed by exonuclear activity (38). The extinction rate due to base G will increase as the amount of base G in the nucleotide sequence of the probe is increased (46).
After the initial and final analysis, it was obtained 3 best mutant probes as TaqMan probe, e.g. K315MT1 mutant probe, K315MN5 and K315MN23. The results of the analysis of the three probes with software can be seen in Table 3. The three TaqMan mutant probes can be used for detection of mutations at the codon of 315 KatG Mycobacterium tuberculosis genes. The criterion values of the three mutant probes (Table 3) have met the general criteria of the probe as described. Although the K315MN5 mutant probe (Table 3) contains more than 3 bases of G and C on 3 bases at the 3 'end, this does not affect the probe because the nucleotide sequence of the probe will not be elongated at the 3' end such as the primer. Therefore, this criterion is very important in the primary design so there is no non-specific elongation process (38). The three mutant probes (Table 3) have also met the end-stage criteria so that the label fluorescence efficiency is not reduced due to the nucleotide sequence of the probe.
Table 3. Final Analysis of Probe Mutan
|
Probe |
Nucleotide sequence o?Probe 5,→3, |
DNA Probe Design Criteria Ai ,⅛⅛a | |||||||
|
Eis |
Tm |
⅞ GC |
Dmer |
fiatrpir, |
runs |
repeat |
Base Amosnt C:G | ||
|
Probe design of specific mutation S315T | |||||||||
|
K315MΓl |
CCACCGGCATCGAGGrCGTATG |
U 4 |
2 | ||||||
|
Probe design of specific mutation S315N | |||||||||
|
K315MN5 |
Atcaccaacggcatcgaggtcg |
59 |
4 |
1 |
73 | ||||
|
K315MN23 |
Caccaacggcatcgaggtcgtat |
23 |
.,0 |
56 |
4 |
7:6 | |||
Information: The probe name, for example K315MT1 shows: M. tuberculosis katG genes; 315 = target codon; M = type of probe is a mutant probe; T = amino acids formed by mutation of codon 315; number 1 = serial number of the
probe design; Pjg = length; Tm = melting temperature (ºC); % GC = percentage of guanine and cytosine base content; underscore = mutation at codon 315.
The probe probe design of 3 probes (Table 3) can be attached to labels. The label for TaqMan probe consists of reporter and quencher. The reporter label is a fluorescent label and placed on the 5 'probe. While the quencher can be fluorescence or non fluorescence compound that absorbs fluorescence from the reporter at the appropriate distance. Quencher can be placed in the center or 3 'probe end (38). The labels commonly used for TaqMan probes are FAM as reporter and TAMRA as quencher (15).
Installation of labels for the design of K315MT1 mutant probes with base changes G → C which marked with bold prints are shown in Figure 8. The reporter label can be embedded on base C at the 5 'end and the quencher label is affixed to a T base at the 3' probe end. Meanwhile, the labeling of the K315MN5 and K315MN23 mutant probe designs is shown in Figure 9. Both designs of this mutant probe have different nucleotide length and compositions, but the same detected nucleotide changes, ie G → A. The type of FAM reporter label can be used on K315MN5 and K315MN23 mutant probes as in Figure 9.
Qj→g⊂g ate ace age ggc ate gag gee gta egg acg aac ace,,.
Q→312 313 314 315 316 317 318 319 320 321 322 323 324...
Kcteraugaie
-
(7) : Kodon genPsrGM IMberciAiosfi Cl)' ^uKle°dda pro⅛a mutaa K-IleMTi
Q: Nuldeodda Hildtype penyusun kodon
Figure 8. Labeling of K315MT1 probe design
Caption:The K315MT1 probe is specific to G → C changes at codon 315, and can be paired with the FAM-TAMRA label
If the K315MN5 or K315MN23 mutant probe will be used in one reaction with the K315MT1 mutant probe, the reporter label of both mutant probe types should be differentiated. The use of reporter labels that provide different fluorescence colors for the S315N and S315T mutations will increase the detection specificity against base changes at codon 315. FAM reporter labels (green fluorescence) can generally be replaced by the VIC label
(fluorescent yellow fluorescence) due to fluorescence from both of these reporter labels typically can be extinguished by TAMRA as a quencher so that no quencher type replacement (30) is required. However, replacement of the label types used should also consider the availability of real-time PCR tools (15, 31).
The selected reporter and quencher labels were then affixed at opposite ends to the nucleotide sequence of the mutant probe (28). Figure 9 shows the label position for the K315MN5 probe (Figure A), with the FAM reporter affixed to the nucleotide A at the 5 'probe end. While the K315MN23 probe reporter (Figure B) is attached to the C nucleotide at the 5 'end of the probe. Both designs of this mutant probe use TAMRA as quencher. The position of TAMRA attachment to the K315MN5 probe is the C nucleotide at the 3 'probe end. Meanwhile, TAMRA for K315MN23 mutant probe is attached to the T nucleotide at the 3 'probe end.
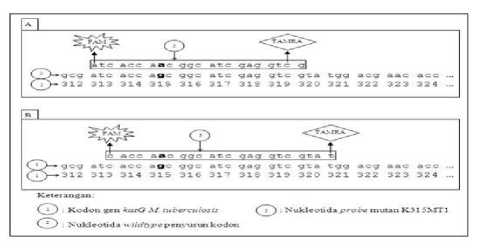
Figure 9. Labeling of mutant probe design for S315N mutation detection
Caption:both K315MN5 and K315MN23 best probes specific to G → A mutations; (A) K315MN5 mutant probe with FAM-TAMRA label, (B) K315MN23 mutant probe with FAM-TAMRA label
The mutant probes that can be labeled based on silico design research analysis with Clone Manager Suite 6 were K315MT1, K315MN5, and K315MN23 probe. The nucleotide sequence of these probes can recognize mutations in the codon of 315 KatG Mycobacterium tuberculosis genes. Specific detection can be produced primarily specific serine mutations into asparagin at codon 315 with K315MN5 and K315MN23 mutant probes, and serine specific mutations into threonine at codon 315 with K315MT1 mutant probe. Therefore, the K315MT1 mutant probe can be used for detection of the S315T mutation. The use of DNA probe detection can provide faster results than conventional PCR and sequencing techniques, as well as cross-contamination and error due to numerous testing stages can be reduced as the amplification and detection process with DNA probe can be performed simultaneously with real-time PCR tool (16). If the test uses two types of mutant probes, such as K315MT1 and K315MN5 probes, the S315T mutation in the tested isolate will cause the K315MT1 probe to be hybridized with specific nucleotide sequence to the target. Then, TaqMan probe K315MT1 will be hydrolyzed and generated fluorescence signal as a marker of S315T mutation. The use of K315MN5, K315MN23 and
K315MT1 mutant probes for mutation detection at codon 315 can provide false-positive results in the presence of mutations in other codons and presence of wild-type isolates. However, further experimental testing is required in the laboratory to prove the performance of the K315MN5, K315MN23 and K315MT1 TaqMan designs.
-
IV. CONCLUSION
-
1. The best mutant probe that can be used for the detection of mutations at the codon of 315 Mycobacterium tuberculosis katG genes is a K315MT1 mutant probe with the nucleotide sequence 5'-FAM-CC ACC GGC ATC GAG GTC GTA TG-TAMRA-3 ' for detection of specific S315T mutations as well as probe mutant K315MN5 and K315MN23 with nucleotide sequences 5'-FAM-ATC ACC AAC GGC ATC GAG GTC G-TAMRA-3 'and 5'-FAM-C ACC AAC GGC ATC GAG GTC GTA T-TAMRA-3' for detection of specific mutations S315N .
-
2. There were 3 DNA probe designs, such as K315MT1, K315MN5, and K315MN23 of the 11 probes that met the initial probe with TaqMan probe criteria for real-time PCR based on minimum base G positions on the 3rd base from tip 5 the probe and the amount of base G does not exceed the base C in the nucleotide sequence of the probe.
REFERENCES
-
[1] World Health Organization(WHO). 2014. Global Tuberculosis Report 2014. Perancis: World Health Organization.
-
[2] World Health Organization(WHO). 2015. Global Tuberculosis Report 2015. Perancis: World Health Organization.
-
[3] Lisdawati, V., N. Puspandari, L. Rif’ati, T. Soekarno, M, Melatiwati, K. Syamsidar, L. Ratnasari, N. Izzatun, dan I. Parwati. 2015. Molecular Epidemiology Study of Mycobacterium tuberculosis and Its Susceptibility to Anti-TuberculosisDrugs in Indonesia. BMC Infectious Disease. Volume 15: 366-373.
-
[4] Babamahmoodi, F., M. R. Mahdavi, H. Jalali, B. Talebi, P. Roshan, dan M. Mahdavi. 2014. Evaluation of Gene Mutations Involved in Drug Resistance in Mycobacterium tuberculosis Strains Derived from Tuberculosis Patients in Mazandaran, Iran, 2013. Int J Mol Cell Med Summer. Volume 3(3): 191-195.
-
[5] Silva, P. E. A. D., dan J. C. Palomino. 2011. Molecular Basis and Mechanisms of Drug Resistance in
Mycobacterium tuberculosis: Classical and New
Drugs. Journal of Antimicrobial Chemotherapy. Volume 66(7): 1417-1430.
-
[6] Tseng, S. T., C. H. Tai, C. R. Li, C. F. Lin, dan Z. Y. Shi. 2015. The Mutations of katG and inhA Genes of Isoniazid-Resistant Mycobacterium tuberculosis Isolates in Taiwan. Journal of Microbiology,
Imunology and Infection. Volume 48: 249-255.
-
[7] Murray, J. L., P. Hu, dan D. A. Shafer. 2014. Seven Novel Probe System for Real-Time PCR Provide Absolute Single-Base Discrimination, Higher Signaling, and Generic Components. The Journal of Molecular Diagnostics. Vol. 16 (6): 627-638.
-
[8] Suryadi, T. 2013. Identifikasi Mutasi Gen katG Pada Isolat P10 dan 151 Mycobacterium tuberculosis Multidrug Resistance di Bali Dengan Metode Polymerase Chain Reaction. Skripsi. Universitas Udayana, Bukit Jimbaran: 1-39.
-
[9] Deniariasih, N. W. 2013. Deteksi Mutasi Pada Gen katG (Fragmen 0,7 kb) Isolat 86 dan P11 Mycobacterium tuberculosis Multidrug Resistance Dengan Teknik Polymerase Chain Reaction (PCR). Skripsi. Universitas Udayana, Bukit Jimbaran: 1-39.
-
[10] Hazbon, M. H., M. Brimacombe, M. B. D.Valle, M. Cavatore, M. I. Guerrero, M. V. Basil, H. B. Jacobe, C. Lavender, J. Fyfe, L. G. Garcia, C. I. Leon, M. Bose, F. Chaves, M. Murray, K. D. Eisenach, J. S. Osornio, M. D. Cave, A. P. D. Leon, dan D. Alland. 2006. Population Genetics Study of Isoniazid Resistance Mutations and Evolution of Multidrug-Resistant Mycobacterium tuberculosis. Antimicrobial Agents and Chemotherapy. Volume 50(8): 2640-2649.
-
[11] Bostanabad, S. Z., L. P. Titov, A. Bahrmand, S. A. Nojoumi. 2008. Detection of Mutation in IsoniazidResistant Mycobacterium tuberculosis Isolates From Tuberculosis Patient in Belarus. Indian Journal of Medical Microbiology. Volume 26(2): 143-147.
-
[12] Jagielski, T., Z. Bakuta, K. Roeske, M. Kaminski, A. Napiorkowska, E. A. Kopec, Z. Zwolska, dan J. Bielecki. 2014. Detection of Mutations Associated With Isoniazid Resistance in Multidrug-Resistant Mycobacterium tuberculosis Clinical Isolates. J. Antimicrob Chemother. Volume 69(4): 2369-2375.
-
[13] Navarro, E., G. S. Heras, M. J. Castano, dan J. Solera. 2015. Real-Time PCR Detection Chemistry. Clinica Chimica Acta. Volume 439: 231-250.
-
[14] Espy, M. J., J. R. Uhl, L. M. Sloan, S. P. Buckwalter, M. F. Jones, E. A. Vetter, J. D. C. Yao, N. L. Wengenack, dan J. E. Rosenblatt. 2006. Real-Time PCR in Clinical Microbiology: Applications for Routine Laboratory Testing. Journal of Clinical Microbiology Review. Volume 19(1): 165-256.
-
[15] Dorak, M. T. 2006. Real-Time PCR. New York: Taylor and Francis Group.1-30.
-
[16] McPherson, M., dan S. Moller. 2006. PCR Edisi 2. New York: Taylor & Francis Group. 209-305.
-
[17] Walker, J.M. dan Rapley, R. 2008.Medical Biomethods Handbook.Totowa, New Jersey: Humana Press Inc. 13341.
-
[18] Wada, T., S. Maeda, A. Tamaru, S. Imai, A. Hase, dan K. Kobayashi.2004. Dual-Probe Assay For Rapid Detection of Drug-Resistant Mycobacterium tuberculosis by Real-Time PCR. Journal of Clinical Microbiology. Volume 42(11): 5277-5285.
-
[19] Kurreck, J. dan C. A. Stein. 2015. Molecular Medicine. Jerman: Wiley-VCH. 37-51.
-
[20] Chou, C.C., C. C. H. Chen, T. T. Lee, dan K. Peck. 2004. Optimization of Probe Length and The Number of Probes Per Gene for Optimal Microarray Analysis of Gene Expression. Nucleic Acid Research. Volume 32(12): 99-107.
-
[21] Kalendar, R., D. Lee, dan A. H. Schulman. 2011. Java Web Tools for PCR, In Silico PCR, and Oligonucleotide Assembly and Analysis. Genomics. Volume 98: 137-144.
-
[22] Yilmaz, L. S., S. Parnerkar, dan D. D. R. Noguera. 2011. mathFISH, A Web Tool That Uses Thermodynamics-Based Mathematical Models for In Silico Evaluation of Oligonucleotide Probes for Fluorescence In Situ Hybridization. Applied And Environmental Microbiology. Volume 77(3): 1118
1122.
-
[23] Zhang, Y. dan W. W. Yew. 2009. Mechanisms of Drug Resistance in Mycobacterium tuberculosis. Int. J. Tuberc Lung Dis. Volume 13(11): 1320-1330.
-
[24] Lina, M., B. Bela, dan A. Yasmon. 2009. Deteksi Mutasi Gen katG Mycobacterium tuberculosis Dengan Metode PCR (Polymerase Chain Reaction)-
Hibridisasi Dot Blot Menggunakan Pelacak Oligonukleotida Bertanda 32P. Jurnal ilmiah Aplikasi Isotop dan Radiasi. Volume 5 (1): 54-67.
-
[25] Smith, J. E. 2009. Biotechnology 5th Edition. New York: Cambridge University Press. 41-44.
-
[26] Anonim a. 2004. Assay Formats for Use in Real-Time PCR. Technical Note. Jerman: Roche Applied Science. 1-13. Available at: https://lifescience.
roche.com/wcsstore/RASCatalogAssetStore/Articles/A ssay%20Format%20for%20use%20in%20Real-Time%20PCR.pdf. (Cited March 17, 2016).
-
[27] Alvandi, E. dan F Koohdani. 2014. Zip Nucleic Acid: A New Reliable Method To Increase The Melting Temperature of Real-Time PCR Probes. Journal of Diabetes & Metabolic Disorders. Vol. 13 (26): 1-4.
-
[28] Livak, K. J., S. J. A. Flood, J. Marmaro, W. Giusti, dan K. Deetz. 1995. Oligonucleotides With Fluorescent Dyes At Opposite Ends Provide A Quenched Probe System Useful For Detecting PCR Product and Nucleic Acid Hybridization. Genome Research. Vol. 4: 357362.
-
[29] Anonim d. 2001. Allelic Discrimination Using The 5’ Nuclease Assay. USA: Applied Biosystems. 1-8. http://www.austincc.edu/mlt/mdfund/mdfund _Unit11AllelicDiscrimination.pdf. (Cited March 14, 2016).
-
[30] Logan, J., K. Edwards, dan N. Saunders. 2009. RealTime PCR: Current Technology and Applications. London: Caister Academic Press. 1-27. Available at:www.horizonpress.com/realtimepcr. http://citeseerx. ist.psu.edu/viewdoc/download?doi=10.1.1.261.223&re p=rep1&type=pdf. (Cited April 23, 2016).
-
[31] Anonim b. 2006. Real-Time PCR Applications Guide. Bulletin 5279. USA: Bio-Rad Laboratories Inc. 1-100. Available at: www.bio-rad.com/webroot/
web/pdf/lsr/literature/Bulletin_5279.pdf. (Cited
February 15, 2016).
-
[32] Holland, P. M., R. D. Abramcon, R. Watson. Dan D. H. Gelfand. 1991. Detection of Spesific Polymerase Chain Reaction Product By Utilizing The 5’→3’ Exonuclease Activity of Thermus aquaticus DNA Polymerase. Proc Natl Acad Sci Biochemistry. Vol. 88: 7276-7280.
-
[33] Liu, H., H. Wang, Z. Shi, H. Wang, C. Yang, S. Silke, W. Tan, dan Z. Lu. 2006. Taqman Probe Array For Quantitative Detection of DNA Targets. Nucleic Acids Research. Vol. 34(1): 1-8.
-
[34] Handoyo, D., dan A. Rudiretna. 2000. Prinsip Umum dan Pelaksanaan Polymerase Chain Reaction (PCR).Unitas. Vol. 9 (1): 17-29.
-
[35] Patel, N. K., dan N. Prakash. 2013. Principle and Tools For Primer Design. Atmiya Spandan Biological Sciences. Vol. 1 (1): 79-95.
-
[36] Bishop, J. L., S. A. Campbell, P. Farrell, M. Fitzgerald, M. Haugen, W. Kocmond, D.E. Madden, W. E. Murray, dan D. H. Persing. 2015. Designing Real-Time Assays on the SmartCycler® II System. United State: Cepheid Technical Support. 1-8.Available at: http://www.cepheid.com/us/ component/phocadownload/category/2-support?download=8:smart-note-6-1. (Cited April 20, 2016).
-
[37] Proudnikov, D., V. Yuferov, Y. Zhou, K. S. Laforge, A. Ho, M. J. Kreek. 2003. Optimizing Primer-Probe Design for Fluorescent PCR. Journal of Neuroscience Methods. Vol 123: 31-45.
-
[38] Lazaro, R. D. dan M. Hernandez. 2013. Real-Time PCR in Food Science Current Technology and Applications. Norfolk, UK: Caister Academic Press. 512.
-
[39] Mulle, J. G., V. C. Patel, S. T. Warren, M. R. Hegde, D. J. Cutler, dan M. E. Zwick.2010. Empirical Evaluation of Oligonucleotide Probe Selection for DNA Microarrays. Plos One. Vol. 5 (3): 1-7.
-
[40] Anonim c. 2006. Beacon Designer 5.10 Manual. Corona Way: USA: Premier Biosoft International. 2289. Available at:
http://211.69.128.172/tlli/upfile/doc/7584c8_Beacon% 20Designer%20510Manual.pdf. (Cited February 25, 2016).
-
[41] Kubista, M., J. M. Andrade, M. Bengtsson, A. Forootan, J. Jonak, K. Lind, R. Sindelka, R. Sjoback, B. Sjogreen, L. Strombom, A. Stahlberg, dan N. Zoric. 2006. The Real-Time Polymerase Chain Reaction. Molecular Aspects of Medicine Review. Vol. 27: 95125.
-
[42] Yuwono, T. 2008. Biologi Molekuler. Jakarta: Penerbit Erlangga. 49-74.
-
[43] Borah, P. 2011. Primer Designing For PCR.Sci Vis. Vol. 3: 134-136.
-
[44] Rychlik, W. 2008.Oligo Primer Analysis Software Version 7. USA: Molecular Biology Insights Inc. 90123.
-
[45] Nazarenko, I., R. Pires, B. Lowe, M. Obaidy, dan A. Rashtchian. 2002. Effect of Primary and Secondary Structure of Oligodeoxyribonucleotides on The Fluorescent Properties of Conjugated Dyes. Nucleic Acids Research. Volume 30 (9): 2089-2195.
-
[46] Mackay, I. M. 2007. Real-Time PCR in Microbiology From Diagnosis to Characterization. Norfolk, Uk: Caister Academic Press. 1-64
Discussion and feedback