Retromolar Embryonal Rhabdomyosarcoma: A Case Report
on
CASE REPORT
RETROMOLAR EMBRYONAL RHABDOMYOSARCOMA: A CASE
REPORT
Caryl Augustine Johanna1, Gede Budhi Setiawan2
1General Surgery Training Programme, Faculty of Medicine, Udayana University, Sanglah General Hospital, Denpasar, Indonesia. Correspondence: caryl_xp@yahoo.com
2Oncology Surgery Subdivision, Surgery Department, Faculty of Medicine Udayana University, Sanglah General Hospital, Denpasar, Indonesia.
ABSTRACT
Background: embryonal rhabdomyosarcoma is common of rhabdomyosarcoma, usually in 5 years old child. Approximately 28% of embryonal rhabdomyosarcomas occur in head and neck area, and 0.04% of cases occur as intra-oral tumors. Case: a 13 years old female complained of a firm and painless progressive mass in her right mandibular retromolar 6 months prior to her current medical check-up. There was a reddish 8x5 cm mass on the right posterior mandibular region. Mid face CT scan showed a well-bordered solid mass in her right oral cavity expanding to the right maxilla without bone destruction nor intracranial expansion. Incisional biopsy concluded the morphology as an embryonal rhabdomyosarcoma. Four series of neoadjuvant chemotherapy of vincristine, adriamycin, and cyclophosphamide (VAC) regimen gave partial response clinically and was followed by right marginal mandibulectomy procedure. Histopathological surgical specimen examination showed no active cancer cells. Adjuvant chemotherapy was given afterward. The result is good cosmetically and functionally. Discussion: retromolar embryonal rhabdomyosarcoma is diagnosed by physical examination of progressive and firm retromolar mass, retromolar mass with or without nearby-structures invasion radiologically, and confirmed by histopathology examination. Neoadjuvant chemotherapy showed partial response clinically. Complete tumor resection with adequate surgical margin is the key for a successful therapy. Conclusion: embryonal rhabdomyosarcoma is rare. The treatment plan and outcome are unique in every patient depending on the location of the tumor, the histological type, and the respectability. Pathological examination and CT scan play a significant role in diagnosing and making a good surgical plan. Early diagnosis and complete resection with adequate surgical margin is the key for a successful therapy.
Keywords: rhabdomyosarcoma, embryonal, retromolar.
RABDOMIOSARKOMA EMBRIONAL RETROMOLAR: LAPORAN KASUS
Caryl Augustine Johanna1, Gede Budhi Setiawan2
1Program Studi Ilmu Bedah, Fakultas Kedokteran Universitas Udayana, Rumah Sakit Umum Pusat Sanglah, Denpasar, Indonesia.
2Subdivisi Bedah Onkologi, Program Studi Ilmu Bedah, Fakultas Kedokteran Universitas Udayana/Rumah Sakit Umum Pusat Sanglah, Denpasar, Indonesia.
ABSTRAK
Latar belakang: rabdomiosarkoma embrional merupakan tipe yang paling sering ditemukan dari jenis rabdomiosarkoma yang mengenai anak berusia 5 tahun. Sekitar 28% angka insiden rabdomiosarkoma terjadi pada regio, kepala dan leher serta 0,04% insidennya timbul sebagai tumor intraoral. Kasus: perempuan usia 13 tahun datang dengan keluhan timbul massa padat yang semakin membesar tanpa disertai rasa nyeri pada area retromolar sejak 6 bulan yang lalu. Terdapat massa berukuran 8x5 cm berwarna kemerahan pada regio mandibular posterior sebelah kanan. Pada
pemeriksaan mid face CT scan menunjukkan gambaran suatu massa solid dengan batas yang tegas pada kavum oral sebelah kanan yang meluas hingga tulang maksila sebelah kanan tanpa disertai dengan kerusakan tulang ataupun perluasan ke daerah intrakranial. Dari pemeriksaan biopsi insisional dapat disimpulkan bentuk morfologi suatu rabdomiosarkoma embrional. Respon parsial ditunjukkan setelah dilakukan kemoterapi neoadjuvan dengan regimen vincristine, adriamycin, dan cyclophosphamide (VAC) sebanyak 4 seri yang dilanjutkan dengan prosedur mandibulektomi marginal kanan. Pemeriksaan histopatologi dari pembedahan menunjukkan sel kanker yang tidak aktif. Kemoterapi adjuvan diberikan setelah prosedur pembedahan dilakukan dengan hasil yang cukup baik secara kosmetik maupun secara fungsional. Diskusi: diagnosis rabdomiosarkoma embrional retromolar ditegakkan melalui pemeriksaan fisik adanya suatu massa padat dan progresif pada area retromolar, dengan atau tanpa adanya invasi ke jaringan sekitar secara radiologis, serta dikonfirmasi dengan pemeriksaan histopatologi. Secara klinis kemoterapi neoadjuvan menunjukkan respon parsial. Reseksi tumor dengan margin yang adekuat adalah kunci dari keberhasilan terapi. Simpulan: rabdomisarkoma embrional merupakan suatu penyakit yang langka. Rencana terapi dan hasil dari setiap pasien berbeda-beda tergantung dari lokasi tumor, tipe histologinya, dan bisa atau tidaknya tumor tersebut dilakukan reseksi. Pemeriksaan patologi dan CT scan merupakan penunjang yang sangat penting dalam mendiagnosis dan perencanaan pembedahan. Penegakan diagnosis sedini mungkin dan total reseksi dengan margin yang adekuat merupakan kunci keberhasilan terapi dari rabdomiosarkoma embrional.
Kata kunci: rabdomiosarkoma, embrional, retromolar.
INTRODUCTION
Rhabdomyosarcoma (RMS) is a mesenchymal malignant neoplasm which accounts for 6% of all the malignancies in children under 15 years.1 The RMS is third common extracranial solid tumor in childhood after neuroblastoma and Wilms’ tumor. Annual incidence of 4-7 per million children below 15 years old.2 Approximately 28% of RMSs occur in head and neck, and amongst them, 0.04% occur as intraoral tumors.3
The clinical presentation varies with the anatomic site, usually presenting as painless and rapidly growing mass.4,5 They are classified histologically to embryonal, alveolar, botryoid, and pleomorphic. It tends to occur in three regions were the head and neck, upper and lower extremities, and genitourinary tract.5 Head and neck RMS occur in young children, most of them is embryonal histology. The other hand, extremity tumors occur in adolescents and have alveolar histologic subtype.2
Current treatment protocols for RMS are designed to deliver risk-based therapy (low, intermediate, or high) based exclusively on the clinical and pathologic features at the time of initial presentation.6 Due to high recurrence rate and association with distant metastases of RMS, a high-quality plan of treatment plan is essential.1
CASE
The parents of a 13-year-old girl took his daughter to the surgical oncology clinic. Her father was a Chinese, while her mother was a mixed German-Indonesian. She had 6 months’ history of right mandibular gingiva tumor. About 7 months ago, her third molar grew and her dentist made an incision above the tooth. Food tend to slip into the defect and one month later the painless tumor started growing. One month before she was taken to the clinic, her right sub mandible seemed to grow bigger as well. She has a history of biopsies taken. The result of the first biopsy was benign
vascular lesion and the result of the second one was benign odontogenic tumor, likely epulis with inflammation. There was a reddish mass on the right posterior mandibular region, irregularly shaped, sized 8x5 cm, solid, painless, and fixed (figure 1a).
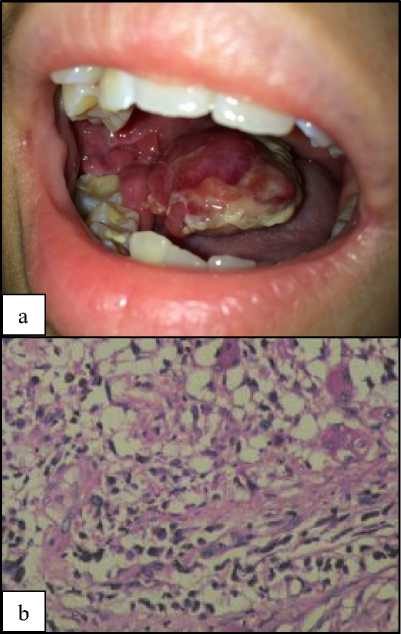
Figure 1. (a) The clinical appearance of the mass intraorally at the first visit of the patient in the surgical oncology clinic. (b) Histopathologically, the tumor was composed of round to spindle cancer cells, some were tadpole shaped, some were cross striated. The nuclei were round to oval, some hyperchromatic nuclei, others had nucleoli.
The initial approach to diagnosis included redo biopsy (figure 1b). The result was round and spindle tumor cells with myxoid background, likely an embryonal rhabdomyosarcoma. The tumor was composed of surface squamous epithelia. There were hypercellular and hypocellular area composed of round to spindle cancer cells, some were tadpole shaped with eosinophilic cytoplasm, some were cross striated. The nuclei were round to oval,
some were uniform. Chromatin were vesicular, some hyperchromatic nuclei, and the others had nucleoli. Mitosis was 36/10. Some of the cancer cells arranged into short fascicular structure. On the hypocellular area, the cancer cells lie between myxoid stroma. Histochemical staining was suggested by the pathologist.
Multisliced computed tomography of head with contrast showed solid mass sized 5x5x8 cm mass in the right oral cavity, narrowing the oral cavity, extended into the right maxilla. There was right and left submandibular glands enlargement (figure 2a). Plain chest x-ray showed no abnormality.
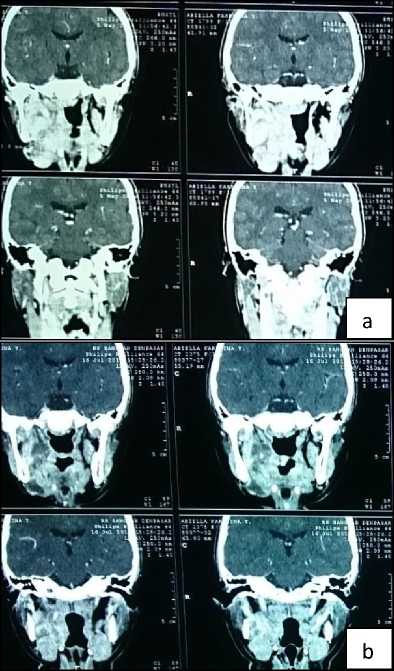
Figure 2. Head CT scan pre (a) and post (b) neoadjuvant chemotherapy. The mass decreased in size.
We decided to give neoadjuvant chemotherapy of vincristine, adriamycin, and cyclophosphamide (VAC) because the tumor was unresectable clinically. After 4
regimens of VAC, there was partial response clinically. Repeated CT scan show decrease of the tumor size, no neck lymphadenopathy, no bone destruction nor intracranial infiltration (figure 2b). Patient was then treated by tumor resection including marginal mandibulectomy (figure 3).
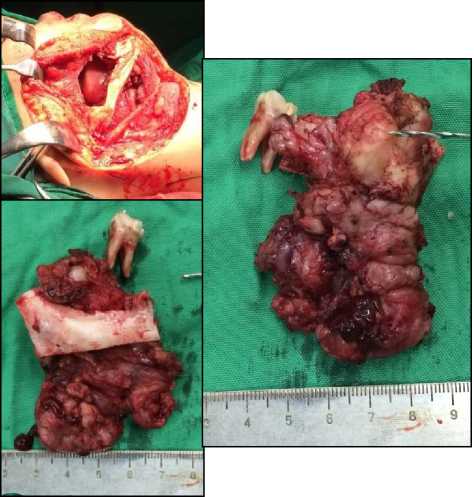
Figure 3. Tumor resection including marginal mandibulectomy was performed after 4 regimens of VAC.
Postoperative histopathology examination showed no active cancer cells. The treatment was followed by VAC regimen as adjuvant chemotherapies. The patient had no functional limitations, though she felt numb on her right mandible and had to check her oral hygiene frequently (figure 4). Regular check-up was an obligation because of the high recurrence evidence.

Figure 4. Picture of the patient before surgery (a) and after surgery (b)
DISCUSSION
Rhabdomyosarcoma is a malignant tumor of mesenchymal origin, arise from cells committed to a skeletal muscle. Annual incidence of RMS is 4-7 per million children below 15 years old. There is a slight male predilection, with ratio male to female is 1.3 to 1.5. The incidence is lower in Asian populations and even lower in African-American than that of the white populations of Western countries.2,4
Rhabdomyosarcomas are classified histologically into embryonal, alveolar, botryoid, and pleomorphic. It occurs in three regions were head and neck, genitourinary, and upper and lower extremities. From these four types, embryonal rhabdomyosarcoma (ERMS) is the most common.5 It is characterized by the loss of heterozygosity at the 11p15 locus, while ARMS (alveolar RMS) are often characterized by a translocation between FOXO1 gene on chromosome 13q14 and either PAX3 on chromosome 2q35 or PAX7 on chromosome 1p36.6
The risk factors of RMS are children below 10 years old but it can also develop in adolescents and adults. It is more common in boys. Some rare inherited conditions can also increase the risk of RMS, such as Beckwith-Wiedemann syndrome, Li-Fraumeni syndrome, Neurofibromatosis type 1, Costello
syndrome, and Noonan syndrome. Some studies have suggested that exposure to x-rays during pregnancy and parental use of drugs such as mariyuana and cocaine might be linked with an increased risk of RMS, but more research is needed to see the true link between these factors and RMS.7 Our patient was a 13 years old female with no familial history of cancer and no known history of x-rays exposure during pregnancy and parental use of drugs. She was mixed Caucasian-Asian. Genetic examination was not performed.
The clinical presentation varies with the anatomic site, usually presenting in less than a year as a painless, rapidly growing mass.4,5 In this case, there was a painless mass, presenting 6 months before the patient came to the surgical oncology clinic, and growing rapidly.
Imaging tests may be done for finding out whether a suspicious area might be cancer, determining the extend of a tumor or learn how far a cancer have spread, and determining if a treatment is working. The results of imaging tests can strongly suggest that someone has RMS, but a biopsy is the only way to be certain. The cells of ERMS look like the developing muscle cells of a 6 to 8 weeks-old embryo.7 That cells have high cytologic variability, representing several stages of skeletal muscle morphogenesis. They can be highly differentiated neoplasms containing rhabdomyoblasts, but more often as poorly differentiated tumor cells.8 Embryonal rhabdomyosarcoma tend to occur in the head and neck area, bladder, vagina, prostate, and testicles.7
We performed biopsy which concluded the mass as an EMRS. The mass, sized 8x5 cm, was fixed to the mandible and submandible. Multisliced computed tomography of head with contrast showed solid mass sized 5x5x8 cm mass in the right oral cavity, narrowing the oral cavity,
extended into the right maxilla. There was right and left submandibular glands enlargement. We concluded the tumor as unresectable, and decided to give neoadjuvant chemotherapy. The VAC regimen is common combination for intermediate-risk group. Irinotecan or topotecan can be added as well. Other drugs used to treat RMS include ifosfamide, etoposide, and doxorubicin.7
Further special stains may be performed to confirm, classify, and determine the prognostic information, such as the Desmin stain, the MyoD1 stain, the PAS stain, the PASD stain, and a Reticalin stain.1,8 Immunohistochemically staining was not performed in this case.
Three important keys to stage RMS are the type of RMS, the TNM stage, and the clinical group. The TNM stage is determined by the tumor, the nodes, and the metastasis before treatment starts. On the other hand, the clinical group is based on the extent of the disease and how completely it is removed during initial surgery.7
Oral RMS is treated with radical excision followed by chemotherapy. It usually using a combination of vincristine, dactinomycin, and cyclophosphamide.1,5 If complete resection is impossible, postoperative radiotherapy can be done. Five years’ survival rates had improved dramatically from less than 10% before the 1960s to 65% today. Stage I lesions had better prognosis (80%). Metastasis can occur through blood or lymphatic vessels. The target organ are cervical lymph nodes, lungs, bones, and brain.5
Complete resection with histologically free margins is often impossible without exenteration, but chemotherapy and radiotherapy can help to complete remission in many cases, so surgery is reserved for unresponsive tumors. Unfortunately, commonly occur
radiotherapy-induced late sequelae.1,5 Head CT scan was repeated after 4 regimens of VAC as neoadjuvant chemotherapy in our case. There was partial response clinically. The surgery afterward (tumor resection including marginal mandibulectomy) concluded complete response in our case. We continued the VAC regimens as adjuvant. The patient had no functional limitations, though she felt numb on her right mandible and had to check her oral hygiene frequently.
By using the 3 keys in staging RMS, we can determine the risk groups of the patients, and hence, the survival rates.7 Our patient has a retromolar ERMS stage I based on the TNM staging and clinical group IIB based on the clinical group stage system, concluding as the low-risk group with over 90% of 5-year survival rate. Most of these children will be cured.7 In general, classic ERMS has a 66% 5-year survival rate. Adverse prognostic factors include adult age, parameningeal location, head and neck location, large size, and unresectability.5,7,9 Regular check-up was an obligation for RMS case because of the high recurrence.
CONCLUSION
Embryonal rhabdomyosarcoma is a rare disease. Pathological examination and CT scan play a significant role in diagnosing and making a good surgical plan. Three important keys to stage and determine the prognosis of RMS are the type of RMS, the TNM stage, and the clinical group. Early diagnosis and complete resection with adequate surgical margin is the key for a successful therapy. Complete resection with histologically free margins is often unfeasible without exenteration, but chemotherapy and radiotherapy enable complete remission in majority of cases.
REFERENCES
-
1. Sahni P, Singhvi A, Nayak MT, et al. Gingival Rhabdomyosarcoma in an Adult: A Unique Entity. Turk Patoloji Derg. 2015;31:153-7.
-
2. Dagher R, Helman L.
Rhabdomyosarcoma: An Overview.
The Oncologist. 1999;4:34-44.
-
3. Patil G, Halawar S, Sagari S, et al. Embryonal Rhabdomyosarcoma
Occuring On Mandibular Gingiva in An Adult. Journal of Clinical and Diagnostic Research. 2013; 7:2088-9.
-
4. Roldan RA, Llanes EGDV, Villarta RL. Embryonal Rhabdomyosarcoma of the Mandible. Phillip J Otolaryngol Head Neck Surg. 2006;21:37-9.
-
5. Tandon A, Sethi K, Singh AP. Oral Rhabdomyosarcoma: A review. J Clin Exp Dent 2012;4:e302-8.
-
6. Chen X, Steward E, Shelat AA, et al. Targetting Oxidative Stress in Embryonal Rhabdomyosarcoma.
Cancer Cell. 2013;24:710-24.
-
7. American Cancer Society.
Rhabdomyosarcoma. (serial online) 2013. [cited 2014 Sep. 01]. Available from: https://old.cancer.org/acs/groups/cid/d ocuments/webcontent/003136-pdf.pdf.
-
8. Ng WKY. Case Report: Embryonal Rhabdomyosarcoma in a Young Boy. McGill J Med. 2007;10:16-9.
-
9. Institut Curie. Childhood cancer: Genetic improves diagnosis and treatment of rhabdomyosarcoma.
(serial online) 2010 Apr. [cited 2014 Sep. 01]. Available from:
http://curie.fr/sites/default/files/rhabdo myosarcoma-genetics-diagnosis.pdf.
52
Discussion and feedback