THE USE OF ADVANCED GENOMIC PLATFORMS TO ACCELERATE BREEDING PROGRAMS OF THE INDONESIAN AGENCY FOR AGRICULTURAL RESEARCH AND DEVELOPMENT (A Review)
on
ARTICLE
THE USE OF ADVANCED GENOMIC PLATFORMS TO ACCELERATE BREEDING PROGRAMS OF THE INDONESIAN AGENCY FOR AGRICULTURAL RESEARCH AND DEVELOPMENT
(A Review)
I Made Tasma
Indonesian Center for Agricultural Biotechnology and Genetic Resources Research and Development (ICABIOGRAD)
Corresponding author : imade.tasma@gmail.com
ABSTRACT
The use of advanced genomic platforms such as next generation sequencing (NGS) and high throughput SNP (HT-SNP) genotyping platform facilitates the use of PGR collection in a more comprehensive manner for a more efficient breeding program. Indonesia is recognized as the second richest mega-biodiversity in the world. This includes the plant and animal GR of agricultural importance. Few excellent example PGR of Indonesian origin included rice, banana, and sweet potato. The available GR diversity richness must be manipulated for human kinds (e.g. breeding purposes) to develop superior crop and animal for food, feed, ornament, and industry. A genomic-based breeding program facilitates the manipulation of the wealth GR collection in a more comprehensive, effective, and efficient manner. This presentation describes the current status of NGS-based genome sequencing project as well as the application of HT-SNP array technology in genomic and breeding projects of the Indonesian Agency for Agricultural Research and Development (IAARD). The main crops and animal under study included soybean, maize, rice, cacao, chili pepper, potato, physic nut, oil palm, and cattle. The sequencing project was done using NGS HiSeq2000 and the SNP chip genotyping study was done using the Illumina iScan. Genome Browser (GB) of the genomic data was developed containing millions of genomic variations (SNP, Indels, and SSR). The GB is an excellent breeding resource to support breeding program of the crop and animal under study. The genome browser also contains phenotypic and SNP genotypic data of specific targeted populations. The SNPs discovered in this study are marker resources for HD SNP chip development. The SNP chips are useful for HT-SNP genotyping projects, GR characterization, and gene tagging to identify superior genes and QTLs for characters of interest to accelerate national breeding program of the national priority crops and animal under study.
Keywords: NGS, genomic variations, SNP chip, germplasm, breeding
INTRODUCTION
NGS technology revolutionizes genomic studies of various plant and animal species. This technology has very low cost of per base sequencing and provide excessisively large amount of sequence data (Ansorge 2009; Varshney et al., 2009). The three main stream of NGS technologies
available involve Roche/454 pyrosequencing platform, the Illumina/Solexa polymerase synthesis sequencing system, and ABI/SOLID ligase technology. Other more recent sequencing platforms included HeliScope, IonTorrent and SMRT (Single Molecule Real Time Sequencing). These latter platforms are able to sequence the
single molecule DNA or RNA. The genetic platforms are very useful in sequencing the plant and animal species of agricultural importance (Gao et al., 2012). Many agriculturely important crops and animals have been sequenced their whole genome and their reference genome sequences have been available (Dhanapal et al., 2011; Van et al., 2012). The availability of the reference genome sequences would ease PGR chacterization and evaluation through genome re-sequencing projects using NGS platforms. For other important crops mainly the tropical ones such as durian, mangosteen, coconut, have not been yet available their reference genome sequences that need to be done. The sequencing projects using NGS result huge number of data and face big challenging for scientists to analyze such data to obtain the genes and their respective markers to be used by plant breeders. Sufficient bioinformatic tools and analyses to be able to use such huge data for discovering genes and their respective markers useful for crop and animal improvement programs are required. In addition, the huge genomic data resulted from such technologies require high capacity computer with high computing power, well-managed data back-up system, various computer and genetic analysis softwares, genome browsers, and multipurpose data bases for handling the genomic and phenotypic data resulted from such crop genomic analyses. This manuscript describes the roles of advanced genomic platforms in accelerating breeding of national priority crops and animal in the crop and animal improvement programs.
Indonesia is rich in plant and animal genetic resources diversity to discover unique and superior genes for crop and animal breeding purposes
As a tropical country located near the equator, Indonesia is one of the countries in the world that are rich in mega biodeversity. Indonesia has been ranked as the second richest biodiversity in the world. Many plants and crop plants cultivated in Indonesia as well as animal species are of Indonesian origin. The plant genetic resources (PGRs) of Indonesia origins include rice, sweet potato, banana, durian, and mangosteen. The animal species of Indonesian origins included various species of poultry. The diversity of the plant and animal GRs is very important resources for developing superior crop varieties and superior animal to feed the Indonesian people and sharing the foods to the world.
The available advanced genomic tools would facilitate PGR characterization to identify and isolate genes and QTLs of agronomic and economic importance. The advanced genomic tools and platforms will exploit the PGRs even faster and characterize the GR in a more comprehensive and more efficient manners in determining and isolating the genes and QTLs of interest (Upadhyaya et al., 2014). The NGS provides tools to sequence whole genome of various tropical crop and animal species including the under utilized crops and animal unique to tropical countries. This includes the construction of genome reference sequences of Indonesian unique crops through de novo sequencing method. The genome reference sequences require hard efforts to be accomplished in tropical crops. Once the reference genome sequences available for particular crop or animal species, the resequencing projects will be used to identify genome variations and isolate and tag superior genes and QTLs of economically important traits. SNP collection from the resequence analyses are resources for SNP chip marker development
to be used for high throughput SNP genotyping in an association study to tag genes and QTLs of traits of interests. The genes and markers are used in molecular breeding using MAS, MAB, and genomic selection (GS). The huge data resulted from advanced genomic analyses of national important priority crops demand super computer and data management system to make data available for scientists and be useful for geneticists and breeders. Bioinformatic analyses become a big challenge to make the genomic data meaningful and be useful for plant and animal breeders in their breeding programs. Molecular breeding will expidate breeding programs to obtain superior varieties of high priority crop and animal species.
Advanced genomic platforms and their applications in Indonesia
At earlier days Sanger sequencing technology was powerful. This method was able to result about 1 kb long of sequence data in one running the platform (Sanger et al., 1997). The sequencing throughput of the Sanger technology, however, was very low making the whole genome sequencing project initiated with this technology took years to finish involving many institutions across the world and spent millions of dollars to complete the human genome sequence. This has dramatically changed lately with the emergence of next generation sequencing (NGS) technology. The NGS genetic platforms have distributed across genome center around the world mainly exist within developed countries.
In the last five years the availability of of advanced genomic platforms has been emerging in developing countries as well, including Indonesia (Tasma 2014a). There are several NGS platforms that have been available at different Indonesian public
research institutes. This included two Illumina Genome Analyzer IIx, one HiSeq2000, one HiScanSQ, and three MiSeq. The platforms distributed across research institutes nationwide. However, not all of the equipments run well, mainly due to the lack availability of enough fundings to run the equipments properly and continuesly. The equipments have also not been managed well as a service system to support a wide variety of biological research institutions and biological scientists to make the use of the equipments in a cheaper and competitive manners.
There are several types of NGS platforms vailable in the market so far, among those, are 454 Roche, Illumina HiSeq2500, and Solid 5500xl (Van et al., 2013) (Table 1). The scientist have loose choices of NGS platforms appropriate for their research interest and research objectives.
With development of the NGS technology, the sequencing cost per unit data sequence has been drastically decreased making the genome sequencing studies becomes more affordable by most scientists around the world not only those who work in developed countries but also those who work and doing genomic research in developing countries. In 2001, the sequencing cost reached 10,000 USD to sequence one megabase (Mb) sequence data compared to abou 1,000 USD to result the same amount of sequence data in 2007, and only about 0.10 USD in 2011 (Figure 1).
This happened because of the rapid development of the NGS technology. With this situation, more and more laboratories are able to conduct whole genome sequencing of their genetic materials to obtain more comprehensive and more meaningful data of their genetic and breeding materials to be
used in genetic analysis and breeding programs.
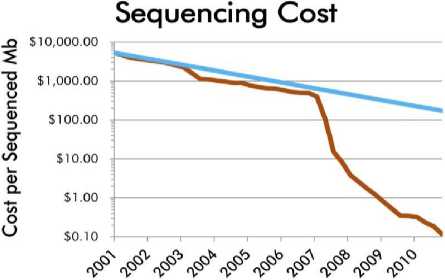
Fig. 1. The actual sequencing cost (orange line) compared to the predicted sequencing cost based on Moor’s law (blue line). Data were obtained from National Human Genome Research Institue (Wetterstrand, 2011).
These genetic platforms have been used to de novo whole genome sequences of crop species of which their reference genome sequences have not yet been available, to whole genome re-sequence of national priority crops of which their reference genome sequences have been available, and for transcriptome sequencing for gene expression analysis purposes, and also for targeted sequencing of specific chromosomal regions.
NGS expidate reference genom sequencing and sequence assembly activities. Using the combination of NGS technology and other sequencing technologies there are more than 20 crop and plant species have been available their reference genome sequences (Van et al., 2012).
Table 1. Main characteristics comparison of of the conventional sanger and some of the most currently used next generation sequencing (NGS) technologies and approximate sequencing cost (in US $ per Mbp) (Perez-de-Castro et al., 2012).
|
Sequencing Technology |
Read Length (bp) |
Mbp per run |
Cost ($/Mbp)a |
|
Sanger |
1000 |
0.001 |
3000.00 |
|
454 Roche |
450 |
450 |
66.00 |
|
Illumina Hi-Seq2000 |
100 |
270000 |
0.07 |
|
Solid 5500xl |
50 |
270000 |
0.07 |
a1 Mbp = 1,000,000 bp
IAARD genome projects to facilitate breeding programs of national priority crops and animals
Reference genome (de novo genome sequencing) of important crops
A reference genome is required for each target species for genomics scientists to leverage the power of modern genome technologies in agricultural research. To produce and annotate the first genome sequence of any individual in a species,
chromosomal DNA is broken up into billions of pieces of about 350-150,000 base pairs in length, and a complex set of specialized DNA libraries are produced. The genomic libraries are then sequences to obtain millions of sequence reads (Figure 2). Longer read sequence data are preferred than shorher reads as the longer reads resulted longer contigs and longer scaffolds (Figure 2). Upon the completion of the difficult, laborious, high computer demand, and high skilled bioinformatics analysis of the scattered pieces of genomic sequences a map
of long contiguous chromosomal regions is reconstructed with a de novo sequence assembly computer (Varsney et al., 2009).
The role of bioinformatic analysis is very high here to be able to complete the scattered pieces of DNA sequences to make unique contigious chromaosomal regions of that particular plant genome. A high capacity computer is required to analyze the sequence data to make contigs consisted of overlapping sequence reads. Many computer softwares are also required and are developed to be able to analyze such huge data into simpler assembly. Longer read sequences enable the computer programs to assemble through the tens of thousands of repetitive elements typically present in plant genomes, which in turn results in longer sequence scaffolds. Long sequence scaffolds have more utility to researchers than short sequence scaffolds (Figure 2).
Once lengthy sequence scaffolds have been produced, expressed regions the genome are typically annotated with millions of sequences generated from RNA libraries also using long read sequencing technologies. Similar to de novo genome sequencing studies, a longer read sequencing technology is also preferred over the shorter read sequencing for the construction of the first-draft of the transcriptome, because long reads when assembled are more likely to result in full-length transcripts, which are most useful for gene model construction. High capability bioinformatics analysis will result high quality reference genome sequence.
The IAARD have conducted de novo sequencing of two crop species, oil palm and physics nut (Jatropha curcas) to provide reference genome maps of the two crops (Tasma et al., 2014b). The oil palm genome reference map, however, was published in 2013 (Singh et al., 2013) and the analyzed
sequence data obtained from three oil palm genotypes conducted by IAARD were used as resequenced data to discover genomic variations of the oil palm genomes. SNP and Indel markers have been developed from the sequenced data to support breeding programs of oil palm. Jatropha curcas de novo sequencing project was conducted in collaboration with Seoul National University (Korea) and Kasersart University (Thailand). The J. curcas genome reference map has been completed that becomes a high value resource to support breeding programs of this oil-bearing crop species. Millions of SNPs and Indels have been discovered by aligning the reference sequences with other genotypes of the Indonesian physics nut accessions. These would be very important marker resources for expidating physic nut breeding programs.
Genome resequencing of plant and animal genetic resources (GR)
Once a set of reference genome exists, scientist can re-sequence a second or third individual from the same species using very low-cost short read technologies, the NGS, such as the Illumina HISEQ-2000 platform (Satyawan et al., 2014). Individual reads from a short read project are not necessarily assembled to one another, but rather are mapped back to the reference genome to identify genetic differences that exist between the reference sequence and the sequence of the second individual. In this way a list of differences can be recorded, and these genetic differences are used to explain differences between varieties within a species, such as yield, disease resistance, flowering time, drought and flood tolerance etc.
Re-sequencing projects can be used to survey variation within a species, to
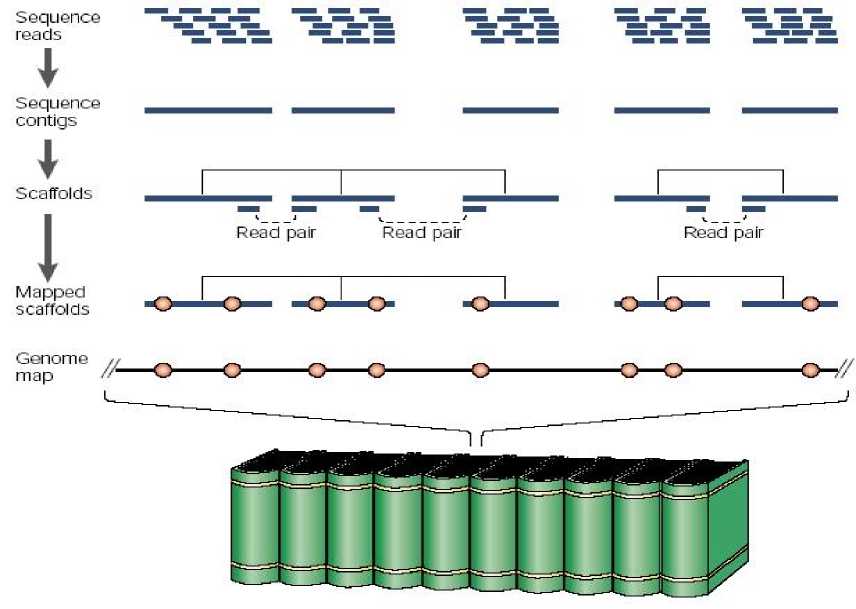
Fig. 2. Schematic diagram of bioinformatic analysis of genome sequence data to build a reference genome sequence of a plant species (Tasma et al., 2012).
conduct genotyping by whole genome sequencing studies, to build high resolution genetic maps, and to generate genome wide association maps (Satyawan et al., 2014).
The role of bioinformatics analysis here is how we can align the resequence data of the selected individual plants to the reference sequence of that particular plant species
From the aligned data then bioinformatics softwares are needed to be able to identify genome variations between the resequence data and the reference genome sequence data. Variations affecting genes can be discovered for isolating gene(s) useful for breeding. The resequencing data can also detect genomic variations (e.g. SNP and INDELs) that are common to all genotypes sequenced and also the ones that are unique to a particular plant genotype analyzed. The roles of bioinformatics and
computational biology are very important here to be able to identify genome variations affecting genes and non gene sequences.
The IAARD has resequenced seven crop species and an animal species (soybean, cacao, oil palm, banana, maize, chili pepper, potato, and beef catlle).
We resequenced breeding parents of the crop species and the Indonesian indigeneous cattle genotypes originated from and unique to different regions of Indonesia. Many genomic variations were obtained from this study (Table 2). The SNPs and Indels obtained from this study are very important resources to support breeding programs of the respective crop and animal species under study.
Table 2. DNA variations observed from alignment of the NGS-derived re-sequence data to the respective reference genome sequence map of the crop and animal species under study by the IAARD (Tasma et al., 2014b; Tasma et al., 2015).
|
Crop/animal Species |
Genome size (Mb) * |
Number of genotypes sequenced |
DNA variation frequency observed |
Total DNA variations Observed |
Total | |
|
SNP |
INDEL | |||||
|
Cacao |
430 |
5 |
121 |
2,326,088 |
362,081 |
2,688,169 |
|
Banana |
472.9 |
14 |
57 |
5,159,450 |
571,885 |
5,731,335 |
|
Oil palm |
1,800 |
3 |
197 |
3,032,200 |
303,109 |
3,335,331 |
|
Maize |
2,300 |
4 |
634 |
2,805,145 |
240,526 |
3,247,037 |
|
Soybean |
1,115 |
5 |
288 |
2,690,000 |
459,000 |
3,150,000 |
|
Chili pepper |
2,649 |
6 |
101 |
24,760,787 |
1,316,987 |
26,077,774 |
|
Potato |
844 |
6 |
160 |
4,166,472 |
339,372 |
4,505,844 |
|
Physic nut |
450 |
3 |
1,185 |
92,007 |
Nd |
92,007 |
|
Cattle |
3,500 |
12 |
127 |
19,979,989 |
919,337 |
20,899,326 |
|
Total and range values |
58 |
57-1,185 |
65,012,138 |
4,512,297 |
69,524,435 | |
*Mb = Mega base pairs (106 bp); **The number of genomes sequenced in each crop or animal species;
*** One DNA variation was observed in each of indicated length of DNA (bp) in the genome; nd, no
data are available yet.
Genome browser data bases development and usage
Various genome browser databases are required to store, manage, and use of the genomic data for several scientific purposes. A genome browser is a graphical interface for display of information from a biological database for genomic data. Genome browsers enable researchers to visualize and browse entire genomes (most have many complete genomes) with annotated data including gene prediction and structure, proteins, expression, regulation, variation, comparative analysis, etc. Annotated data is usually from multiple diverse sources. They differ from ordinary biological databases in that they display data in a graphical format, with genome coordinates on one axis and the location of annotations indicated by a spacefilling graphic to show the occurrence of genes, etc. With the development of reference genome and re-sequencing genome
of various plant and crop species, many genome brosers have been developed to be able used by plant scientists discovering particular genes, developing genetic markers, or structural genomics as well as learning various gene structures, expressions and gene regulation. Example of common genome browsers found in the web that can be used regularly by plant scientis is as shown in Table 3.
With the genome browsers plant scientists can use them to find genes in related crop or from unrelated plant species by comparative genomic studies. The roles of bioinformatics in developing and in using the genome browser data bases of various crop species are very impotant to be able to apply such databases for many genomic and breeding activities (e.g. discovering important genes and DNA markers associated with targeted superior traits of breeder interests.
Table 3. Some important databases and repositories of genomic information of interest for breeders (Perez-de-Castro et al., 2012).
|
Database name |
Web site content |
Web site |
|
Genbank |
General public sequence repository | |
|
EMBL |
General public sequence repository | |
|
DDBJ |
General public sequence repository | |
|
NCBI |
Biomedical and genomical information | |
|
Gene Index Project |
Transcriptome repository | |
|
GOLD |
Repository of genomes databases | |
|
Phytozome |
Genomic plant database | |
|
Plantgdb |
Genomic plant database | |
|
CropNet |
Genomic plant database | |
|
SGN |
Solanaceae information resource | |
|
Gramene |
Grass information resource | |
|
MaizeGDB |
Maize infornation resource | |
|
Tair |
Arabidopsis information resource | |
|
CotthonDB |
Cotton information resource |
The IAARD has developed genome browsers covering genomic and phenotypic data of rice, soybean, maize, cacao, oil palm, physic nut, chili pepper, potato and beef cattle. The genome browsers contain genome wide resequencing data of each respective crop and animal species, SNP and Indel information (gene or non-gen SNPs and Indels) across the genome including the sequence information where the SNps and Indels located, etc. The genome browsers are being incorporated into the IAARD open-to-public genome database (http://genom.litbang.pertanian.go.id). This database is a very important resource for crop and animal scientists (geneticists, plant and animal physiologists and breeders) interested in finding particular genes and genetic markers of particular traits of the respective crop and animal species covered
by the IAARD genome database. The genome resources should provide the basic
data for developing breeding resources to expidate national breeding programs of the national priority crop and animal species.
Genomic-based plant breeding to accelerate breeding programs of national priority crop and animal species
In plants, it has been commonly practiced to tag traits with molecular markers. More classical genomic tools have been successfully used to tag many important agronomic traits with RFLP and SSR markers. The markers linked to the traits then are used in a molecular breeding program to select individual plants having the trait of interest. Such marker-assisted selection (MAS), marker-assisted backcrossing (MAB) become very common to be applied in resent years to expidate breeding programs. More recently with the ability to map plant genome with dense molecular markers genomic selection (GS)
technique becomes more popular for plant breeders.
The GS is defined as the simultaneous selection for many thousands of markers, covering the entire genome so that all genes are expected to be in linkage disequilibrium with at least some of the markers (Meuwissen et al., 2001). The GS is very compatible with the resent genotyping technologies using NGS and high throughput SNP genotyping methods in which the marker assay can cover thousands and even few millions SNP markers. This technology becomes more feasible with a drop in genotyping costs, has GS become feasible, attracting the attention of crop and perennial plant breeders (Bernardo and Yu 2007). Genome-wide and cost-efficient marker systems are needed to apply GS in plants. Furthermore, dedicated GS breeding populations with effective population sizes (Ne) of around 20–50 individuals must be adopted to increase the extent of LD and fit into currently achievable genotyping densities of a few hundred markers per Morgan. In genetically heterogenous populations still encompass large amounts of genetic variation for sustained genetic gains, recent experimental results in outbred plants, indicate that GS has better predictive ability than the classical polygenic model (Lee et al., 2008). A GS scheme has been tested in several crop plants where predictive equations for multiple traits are developed on the basis of high density genotyping and precise phenotyping of several hundred invididual plants of a discovery population (training set) involving Ne in the range of 15–50. Selection accuracy of the predictive models is then assessed in a validation population, targeting the application of early GS at the individual plant level in progeny trials. This technique, once accomplished will be very accurate in selecting individual
plants having the trait of interest and can be done in a faster, more predictable, and more precise manner. However, its applications in Indonesia demonstrates more challenges as many of our tropical crop species are genetically less defined than other more common food crops such as rice, maize, and soybean. Plant geneticists needs to provide better genetic information for the crop targets to be able to apply genomic-based breeding technology using the high density molecular marker technology in their breeding programs.
The IAARD genomic-based breeding programs have been conducted on the seven crop species and an animal species of which their genomic characterization have been done recently. A national consorsium was establi-shed in 2010 consisting of commodity-based research intitutes nationwide. The consortium members included Research Institute for Vegetable Crops, Research Instute for Tropical Fruit Crops, Research Intitute for Oil Palm, Research Insitute for Industrial Crops, and Research Insitute for Animal Production, and Research Institute for Biotechnology and Genetic Resources. The present traits under study included drought tolerant for maize, aluminum-toxicity tolerant for soybean, pod rot resistance for cacao, oil yield and quality and slow-stem-growth rate of oil palm, high oil yield and quality of physics nut, and high meat productivity of beef cattle (Tasma et al., 2014b). Genomic-based gene and QTL tagging would be done using segregating populations developed by the respective research institutes before genomic base breeding programs are done by the consortium members to expidate breeding programs of the respective traits in the respective crops and animal under study.
CONCLUSION
Advanced genomic technologies revolutionized plant genomics and crop improvement programs. NGS technology makes it possible to make reference genome sequences of important crop species through de novo sequencing project and accurate and fast plant and animal GR characterization through genome resequencing projects. NGS and high-throughput genotyping technologies would genotype PGR and animal GR in a more comprehensive way with more chance in identifying individual plants or animal having superior alleles of targeted traits. Bioinformatics as well as high capacity computer, softwares, genome browser databases play important roles in hadling, managing, and applying the huge genomic data resulted from advanced genomic technologies to identify and isolate genes and QTLs useful for breeding programs. The genomic-based breeding programs through MAS, MAB, and GS would result superior crop cultivar and superior animal in a faster, more precise, and more effective manners. The IAARD has used the NGS platforms to obtain millions of genome-wide SNP and Indel variations of seven important crop species and an important animal species that are of high value breeding resources for supporting the important priority crop and animal breeding programs. The SNP collections obtained from the study, upon verification, are used in synthesizing high density SNP chips containing thousands of SNP markers to comprehensively characterize PGR and animal PBR and to tag economically important genes and QTLs to be used to expidate breeding programs of the seven crops and an animal species under study.
REFERENCES
Ansorge, W.J. 2009. Next-generation DNA sequencing techniques. Nature Biotechnol. 25: 195–203.
Bernardo, R., and J.M. Yu. 2007. Prospects for genomewide selection for quantitative traits in maize. Crop Science 47: 1082-1090.
Dhanapal, A.P. 2012. Genomics of crop plant genetic resources. Advances in Bioscience and Biotechnology 3: 378-385.
Gao, Q., G. Yue, W. Li., J. Wang, J. Xu, and Y. Yin. 2012. Recent progress using high-throughput sequencing
technologies in plant molecular breeding. Journal of Integrative Plant Biology 54 (4): 215-227.
Lee, S.H. J.H. van der Werf, B.J. Hayes, M.E. Goddard, P.M. Visscher. 2008. Predicting unobserved phenotypes for complex traits from whole-genome SNP data. PLoS Genet 4: p.
e1000231.
Meuwissen, T.H., B.J. Hayes, M.E.
Goddard. 2001. Prediction of total genetic value using genome-wide dense marker maps. Genetics 157:
1819-1829.
Pérez-de-Castro, AM, S. Vilanova, J.
Canizares, L. Pascual, J.M. Blanca, M.J. Diez, J. Prohens, and B. Pico. Application of genomic tools in plant breeding. Current Genomics 13 (3): 179-195.
Sanger, F., S. Nicklen, and A.R. Coulson. 1997. DNA sequencing with chainterminating inhibitors. Proc. Nat. Acad. Sci. U.S.A. 74: 5463-5467.
doi:10.1073/pnas.74.12.5463
Satyawan, D., H. Rijzaani, I.M. Tasma. 2014. Characterization of genomic variation in Indonesian soybean (Glycine max) varieties using nextgeneration sequencing. Plant Genetic Resources 12: S109-S113. DOI:
http://dx.doi.org/10.1017/S14792621 14000380.
Tasma, I.M., D. Satyawan, H. Rijzaani, D.W. Utami, P. Lestari, and I. Rosdianti. 2012. Pembentukan empat peta genetik sawit, jarak pagar, padi, dan kedelai, serta identifikasi marka SNP kakao dan sapi. Laporan Akhir Penelitian APBN 2012. BB Biogen, Badan Litbang Pertanian. 67 pp.
Tasma, I.M. 2014a. Single nucleotide polymorphism (SNP) sebagai marka DNA masa depan. WartaBiogen 10 (3): 7-10.
Tasma, I.M., H. Rijzaani, D. Satyawan, P. Lestari, Reflinur, I. Rosdianti, E. Mansyah, R. Kirana, Kusmana, M. Pabendon, Rubiyo, et al., 2014b. Analisis genom dan sidik jari komoditas pertanian strategis. Laporan Akhir Penelitian 2014. BB Biogen Bogor. 79 pp.
Tasma, I.M., H. Rijzaani, D. Satyawan, P. Lestari, D. W. Utami, I. Rosdianti, R. Purba, E. Mansyah, A. Sutanto, R. Kirana, Kusmana, A. Anggraeni, M. Pabendon, and Rubiyo. 2015.
Next-Gen-Based DNA Marker Development of Several Importance Crop and Animal Species. Presented at
SABRAO 13th Congress and
International Conference 14-15 September 2015, Bogor, Indonesia, 8pp.
Upadhyaya, H.D., S. Sharma, K.N. Reddy, S.K. Singh, S. Singh, R.K. Varshney, and C.L.L. Gowda. 2013.
Utilization of cultivated and wild germplasm in crop improvement. 3rd International Symposium on
Genomics of Plant Genetic Resources 16-19 April, 2013, Jeju, Korea. P.18.
Van, K., K. Rastogi, K.-H. Kim, and S.-H. Lee. 2013. Next-generation
sequencing technology for crop improvement. SABRAO Journal of Breeding and Genetics 45 (1): 84-99.
Varshney, R.K., S.N. Nayak, G.D. May, and S.A. Jackson. 2009. Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 9: 522530.
Wetterstrand, K.A. 2011. DNA
Sequencing Costs: Data from the NHGRI Large-Scale Genome Sequencing Program Available at: http://www.genome.gov/sequencingco sts/. Accessed March 1, 2011.
53 • asia oceania bioscience and biotechnology consortium
Discussion and feedback