Diagnosis dan Tatalaksana Sindrom Treacher Collins
on
ARTIKEL TINJAUAN PUSTAKA

J'W∏ijl
Essence of Scientific Medical Journal (2020), Volume 17, Number 2:9-14 P-ISSN.1979-0147, E-ISSN. 2655-6472
TINJAUAN PUSTAKA
DIAGNOSIS DAN TATALAKSANA SINDROM TREACHER COLLINS
Bertha Kawilarang,1
ABSTRAK
Pendahuluan: Sindrom treacher collins adalah penyakit genetik langka yang ditandai berbagai malformasi kongenital dengan manifestasi klinis terutama di bagian kraniofasial.
Pembahasan: Diagnosis dapat dilakukan sejak periode prenatal atau postnatal, namun analisis genetik merupakan diagnosis definitif untuk menentukan adanya mutasi gen. Hal ini disebabkan oleh etiologi sindrom treacher collins yang didasari kelainan kromosom 5q32-q33.1 dengan mutasi delesi pada gen TCOF1, serta gen lain yang diduga berhubungan yaitu POLR1D dan POLR1C. Sindrom ini diturunkan secara autosomal dominan sedangkan pada sebagian kecil kasus secara autosomal resesif. Diagnosis klinis ditegakkan dari temuan pemeriksaan fisik tipikal yaitu malformasi kraniofasial pada bentuk dan fungsi antara lain mata, telinga, maksila, mandibula dan jalan nafas. Spektrum klinis dari malformasi kraniofasial sangat beragam karena didasari oleh abnormalitas perkembangan tulang dan kartilago dari neural crest cell. Tidak ada terapi yang dapat menyembuhkan sindrom ini, sehingga tatalaksana pasien bertujuan untuk memperbaiki bentuk dan fungsi malformasi kraniofasial sesuai yang pasien alami. Tatalaksana pasien membutuhkan tim multidisiplin dengan perencanaan tindakan operatif maupun non-operatif yang dilakukan sejak lahir sampai usia dewasa. Adanya manifestasi klinis lain seperti gangguan pendengaran dan bahasa dapat menyebabkan keterlambatan tumbuh kembang anak jika intervensi tidak dilakukan sejak dini. Sehingga tidak hanya penanganan dari dokter spesialis bedah plastik, namun perawatan berkelanjutan dari spesialis lain juga penting antara lain spesialis anak, telinga dan tenggorokan, audiologis, maupun oftalmologis.
Kesimpulan: Diagnosis dan penanganan sindrom treacher collins masih merupakan tantangan karena aspek fungsional dan estetik pasien harus dipertimbangkan. Pasien yang telah didiagnosis harus segera dirujuk ke pusat khusus dengan tim kraniofasial yang melibatkan tim multidisipilin.
Kata kunci: Diagnosis, Malformasi kraniofasial, Sindrom Treacher Collins, Tatalaksana
ABSTRACT
Introduction: Treacher Collins syndrome is a rare genetic disease which is marked by various congenital malformation with clinical manifestation mainly in the craniofacial region.
Discussion: Diagnosis can be made since prenatal or postnatal period, however genetic analysis remains as definitive diagnosis to determine gene mutation. This is due to the etiology of treacher collins syndrome which is based on abnormality of 5q32-q33.1 chromosome with deletion mutation at TCOF1 gene, as well as other presumed genes associated which are POLR1D and POLR1C. This syndrome is inherited through autosomal dominant while a minority of case is found through autosomal recessive inheritance. Clinical diagnosis is based from typical physical examination findings which are craniofacial malformation of the form and function of organs namely the eyes, ears, maxilla, mandible and airway. There is a wide clinical spectrum of this craniofacial malformation as it is based on growth abnormality of the bone and cartilage derived from neural crest cells. There is no cure for the syndrome, thus management of the case is aimed to imrpove form and function of the craniofacial malformation according to patient’s symtpoms. Management of patient requires a multidisciplinary team, with planning of operative or non-operative approaches since birth to adult. Presence of other clinical manifestations such as hearing and speech disturbance may cause developmental delay if no early intervention is taken. Hence not only is treatment management needed from plastic surgeon, instead care from other involved specialist is also required such as pediatrician, ENT specialist, audiologist and ophthalmologist.
Conclusion: Diagnosis and management of Treacher Collins Syndrome is still a challenge due to patient’s functional and esthetic aspect that must be fully considered. Patient who has been diagnosed must promptly be referred to specialized center with craniofacial team involving multidisciplinary team.
Keywords: Diagnosis, Craniofacial malformation, Treacher collins syndrome, Management
1Program Studi Pendidikan Dokter, Fakultas Kedokteran, Universitas Pelita Harapan, Tangerang
PENDAHULUAN
Sindrom treacher collins atau disebut juga sindrom Franceschetti–Zwahlen–Klein merupakan suatu penyakit genetik yang langka akibat mutasi pada gen TCOF1. Sindrom ini meliputi kelainan pada kraniofasial, yang disebabkan oleh disgenesis jaringan di arkus brankial satu dan dua.[1] Disgenesis sendiri merupakan suatu perkembangan organ yang abnormal pada masa embriologi. Sindrom ini pertama kali ditemukan oleh dokter spesialis mata dari Inggris, Edward Treacher Collins, yang kemudian dipublikasikan dalam jurnal pada tahun 1900.[2] Namun pada tahun 1949, Fransceschetti dan Klein mengulas kembali paparan mengenai sindrom ini dan menyebutnya suatu disostosis mandibulofasial.[1,2] Disostosis merupakan kelainan pada perkembangan tulang,
terutama pada tahap osifikasi tulang. Umumnya tatalaksana awal sindrom treacher collins meliputi proteksi patensi jalan nafas, fungsional mata, serta perkembangan auditorik dan neurologis.[3]
Selanjutnya tatalaksana kasus sindrom treacher collins mengikuti prinsip bedah kraniofasial.[4] PEMBAHASAN
Epidemiologi
Sindrom treacher collins merupakan kondisi yang diturunkan secara autosomal dominan pada gen TCOF1 di kromosom 5q32.[1] Kelainan pada sindrom ini memiliki karakteristik hipoplasia pada jaringan lunak kraniomaksilofasial dan hipoplasia pada arkus brankial satu dan dua.[1,2] Tingkat insiden sindrom treacher collins diperkirakan 1 dari 25,000 dan 1 dari 50,000 kelahiran hidup.[5] Tidak
ada predileksi pada jenis kelamin tertentu. Sementara itu, sebanyak 60% kasus sindrom treacher collins adalah akibat mutasi de novo.[1,5] Namun belum ditemukan adanya korelasi antara genotip dan fenotip tertentu pada sindrom treacher collins.[5] Pada hampir seluruh kasus, sindrom treacher collins didiagnosis saat lahir, tetapi dengan adanya dismorfologi wajah yang tipikal pada kasus yang berat, kondisi ini dapat didiagnosis lebih awal saat prenatal melalui ultrasonografi. Pada kasus yang lebih ringan, sindrom ini tidak dapat didiagnosis saat lahir karena tidak dikenali sebagai suatu sindrom treacher collins hingga muncul komplikasi lain.[2,5] Sebuah kasus melaporkan gejala retrognatia ringan yang tidak khas dengan sindrom treacher collins sehingga baru dikenali saat usia remaja bersamaan dengan munculnya penurunan pendengaran kronis.[6] Rekonstruksi telinga diperlukan untuk diagnosis pasti anomali pada bagian telinga dan memperbaiki fungsi pendengaran.[6]
Etiologi
Studi genetik menunjukkan bahwa kelainan sindrom treacher collins disebabkan oleh kelainan pada kromosom 5q32-q33.1.[4] Didapatkan mutasi jenis delesi pada gen TCOF1 (Treacle Ribosome Biogenesis Factor 1) yang terdiri atas 26 ekson dan mengkode sebanyak 1411 protein asam amino yang disebut treacle.[4] Kadar ekspresi TCOF1 terbanyak ditemukan pada sel crest of neural folds dan arkus faringeal satu. Adanya mutasi delesi menyebabkan berkurangnya neural crest cells (NCC) yang berperan pada perkembangan embriologis daerah kraniofasial melalui transkripsi gen yang
menghasilkan protein treacle.[2,4] Treacle diperlukan dalam pembentukan dan proliferasi NCC, salah satunya untuk berdiferensiasi sebagai kartilago dan tulang yang merupakan komponen penting kraniofasial.[7] Hal ini didukung oleh studi pada mencit dimana adanya mutasi pada TCOF1 terbukti menurunkan jumlah NCC melalui peningkatan apoptosis.[8,9] Selain itu, pada 9% kasus didapatkan juga mutasi pada gen POLR1C dan POLR1D yang berperan pada transkripsi RNA.[1] Sebuah studi dengan uji hewan model menemukan bahwa mutasi pada POLR1C menyebabkan berkurangnya biogenesis RNA sehingga menginduksi p53 yang meningkatkan kematian sel.[10] Selain itu disfungsi pada POLR1C juga mengganggu pembentukan NCC sehingga terjadi hipoplasia tulang wajah pada malformasi kraniofasial.[10] Beberapa studi melaporkan adanya pola penurunan gen POLR1C secara autosomal resesif.[1,2]
Gejala Klinis
Karakteristik sindrom treacher collins sangat khas dan berbeda dari sindrom lainnya. Hampir keseluruhan kelainan disebabkan oleh kelainan pada arkus faringeal satu dan dua. Kelainan lain dapat meliputi beberapa organ antara lain okular dan periorbital, aurikuler, zigoma, maksila dan mandibula.[5] Dapat dijumpai adanya kelainan tuli maupun anomali pada daerah genital. Umumnya pasien memiliki tingkat intelegensi yang normal.[11]
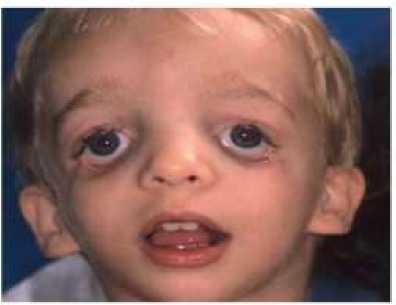
Gambar 1. Karakteristik orbital pada pasien sindrom Treacher-Collins[5]
Okular dan Periorbital
Salah satu ciri khas sindrom treacher collins adalah dismorfisme pada bola mata disertai hipoplasia tulang zigoma dan bentuk ellipsoid.[2] Sebagai akibat dari hipoplasia pada zigoma dan pembentukan struktur dinding lateral bola mata yang kurang baik, bola mata tampak lebih menonjol.[4] Prominensia bagian nasal dan maksila umumnya normal tetapi pada beberapa kasus dapat ditemukan juga enoftalmus.[5] Fisura palpebra yang tampak tertarik ke bawah atau “down-slanting” umumnya menunjukkan suatu presentasi “anti-mongoloid”.[12]
Struktur jaringan lunak di sekitar mata umumnya hipoplastik atau kurang terbentuk. Bagian kelopak mata bawah seringkali tipis dan terdapat lekukan yang disebut koloboma.[1,2] Selain itu, otot orbikularis oris dan kelenjar meibomian juga tidak dapat ditemukan pada pasien sindrom treacher collins.[4] Presentasi klinis yang patognomonis lainnya adalah margin kelopak mata dengan laksitas signifikan, dan hilangnya bulu mata terutama pada bagian sepertiga medial kelopak mata bawah.[5] Selain itu dapat ditemukan atresia duktus lakrimalis dan hilangnya pungta mata.[1,5]
Pada gejala klinis, sebanyak 33% pasien dapat mengalami kebutaan, 37% mengalami strabismus, dan pada sebagian kasus dapat ditemukan katarak kongenital sampai anoftalmia.[1,2] Berbagai kelainan mata tersebut dapat disebabkan oleh kondisi ambliopia oleh adanya jaringan parut pada kornea, strabismus, maupun kelainan refraksi mata yang berat.[1,4] Namun pada sebagian besar penyandang sindrom ini, visus tetap normal karena perkembangan retina tidak melalui arkus brankial, sehingga setidaknya pasien memiliki satu sisi mata dengan visus yang masih normal.[13]
Aurikular
Sebanyak 87% kasus pasien sindrom treacher collins disertai dengan malformasi pada bagian telinga.[14] Gejala klinis yang menonjol adalah deformitas telinga eksternal, yaitu mikrotia bilateral atau anotia. Kelainan ini disertai posisi telinga atau garis rambut yang terletak lebih rendah pada 48% kasus.[14] Selain itu, umumnya ditemukan stenosis atau atresia dari meatus auditorik eksternal. Membran timpani pada bagian dalam telinga juga umumnya memiliki deformitas bentuk.[3,5] Walaupun bagian dalam telinga secara morfologis normal tetapi dari segi fungsionalitas tetap ditemukan adanya tuli konduktif.[13] Derajat tuli konduktif dapat bervariasi pada setiap pasien.[15]
Zigoma
Kelainan pada struktur wajah tengah merupakan gejala klinis lain yang patognomonis pada sindrom treacher collins. Adanya hipoplasia pada bagian zigoma berdampak pada lebar struktur wajah tengah yang lebih pendek.[15] Hal ini diperkuat oleh studi dari Posnick et al yang menunjukkan bahwa melalui foto CT aksial tampak jarak dan panjang arkus zigoma yang lebih pendek.[1,2,16] Derajat deformitas zigoma diklasifikasikan menurut morfologi dan volume. Pada tipe 1 (minor) terdapat seluruh komponen zigoma, tipe 2 (moderat) hanya sebagian komponen zigoma dan tipe 3 (berat) tidak terdapat komponen zigoma.[5]
Maksila dan mandibula
Deformitas mandibula pada pasien sindrom treacher collins sangat bervariasi bahkan dapat mencakup seluruh spektrum hipoplasia mandibula.[16] Dampak dari karakteristik retrognatia yang prominen adalah bentuk wajah pasien yang sangat konveks disertai malformitas pada bentuk mandibula.[17] Sudut mandibula tidak terbentuk secara signifikan bahkan dapat tidak tampak sama sekali.[13,17]
Antegonial notch yang merupakan bagian perbatasan bawah ramus yang menyatu dengan mandibula, tampak lebih tinggi disertai dengan jarak 1/3 bawah wajah yang memendek.[1,2] Bagian ini juga dapat terputar searah jarum jam sehingga menimbulkan bentuk wajah yang sangat konveks.[1,2] Jika dilihat menurut proporsi wajah, maka panjang bagian wajah posterior memendek sedangkan bagian bawah anterior wajah semakin memanjang.[5] Penilaian deformitas mandibula akibat hipoplasia dapat diklasifikasikan menurut modifikasi Kaban/Mulliken.[5] Gambar 2 menunjukkan tipe I dengan bentuk normal, namun mandibula dan TMJ lebih kecil. Tipe IIA terdapat hipoplasia mandibula moderat, hipoplasia ramus, namun perkembangan posisi TMJ normal. Tipe IIB terdapat hipoplasia mandibula moderat-berat dengan hipoplasia ramus dan kondilus, dengan sendi TMJ malposisi inferior, medial dan anterior. Sedangkan pada tipe III tidak ditemukan ramus mandibula.
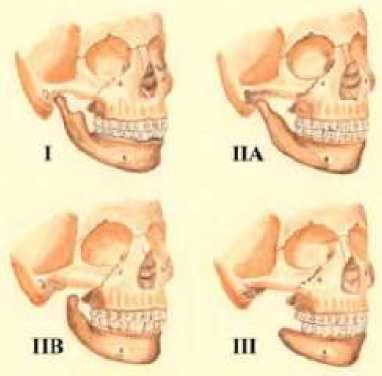
Gambar 2. Klasifikasi Kaban/ Mulliken[5]
Bagian hidung pasien sering digambarkan seperti berbentuk paruh.[11] Hal ini disebabkan oleh hipoplasia pada jaringan sekitar yang menghasilkan suatu ketidakseimbangan wajah.[5] Namun jika
dilihat dari studi antropometrik tidak ditemukan kelainan dari ukuran ataupun bentuk hidung.[5,11]
Abnormalitas pada struktur organ lain juga sering ditemukan seperti atreasia koana, celah palatum bentuk komplit maupun submukosa, tidak terbentuknya kelenjar parotid, malformasi tulang belakang servikal, maupun penyakit jantung kongenital.[18] Akan tetapi kelainan-kelainan tersebut bukan merupakan karakteristik kardinal dalam sindrom treacher collins sehingga tidak selalu ditemukan.[18] Malformasi lain dari sindrom treacher
collins dapat ditemukan melalui beragam pemeriksaan penunjang yang terlihat dalam tabel 1.
Tabel 1. Kriteria diagnosis sindrom Treacher Collins
|
Pemeriksaan Fisik | |
|
Mata |
Fisura palpebra tampak anti-mongoloid, koloboma pada kelopak mata bawah, aplasia bulu mata, hipertelorisme, dakrostenosis |
|
Fasial |
Tidak ada tulang malar, arkus zigomatikus, hipoplasia temporalis, garis rambut rendah, aplasia atau hipoplasia kelenjar parotis |
|
Telinga eksternal |
Mikrotia bilateral, telinga letak rendah, stenosis atau atresia bilateral pada meatus auditorik eksternal |
|
Telinga bagian tengah |
Tuli konduktif, hipoplasia atau agenesis malleus dan inkus |
|
Mulut |
Makrostomia, celah palatum dengan atau disertai celah bibir, inkompetensi velofaringeal, arkus palatum tinggi |
|
Gigi |
Agenesis gigi, opasitas enamel, profil gigi birdlike appearance, maloklusi dan erupsi ektopik dari gigi molar pertama |
|
Saluran nafas |
Hipoplasia mandibula menyebabkan gangguan pernafasan, hipoplasia faringeal, atreasia koana, fistula trakeo-esofagal, obstruksi nasal |
|
Pemeriksaan Penunjang | |
|
Ultrasonogra fi |
Pada trimester dua dan tiga dapat mendeteksi kelainan kraniofasial yang berat |
|
Radiografi sefalometri |
Malformasi pada gigi, hipoplasia mandibula dengan konkavitas yang khas pada bagian bawah mandibula, pada tengkorak tampak copper-beatan appearance |
|
Radiografi water’s view |
Hipoplasia atau aplasia arkus |
|
zigomatikus | |
|
CT scan |
Telinga: Antrum dan atik berbentuk slit-like dengan ukuran lebih kecil dari normal, tidak tampak pneumatisasi pada mastoid, atresia koana dan telinga bagian tengah Regio fasial: Hipoplasia pada arkus zigomatikus dan mandibula dengan protrusio wajah bagian tengah |
|
MRI |
Hipoplasia pada otot masseter dan kelenjar parotis, sinusitis maksillaris dan sfenoidal |
|
Pemeriksaan molekular genetik |
Ditemukan varian patogenik TCOF1, POLR1C atau POLR1D |
Diagnosis
Analisa molekular genetik melalui gene-targeted testing atau genomic testing komprehensif merupakan metode definitif untuk diagnosis sindrom treacher collins secara prenatal atau postnatal.[12] Umumnya tampilan struktur wajah mulai dapat terlihat pada usia kehamilan 30 minggu namun, kemampuan ultrasonografi 2-dimensi terbatas dan tidak cukup untuk menilai profil fetus.[5] Penggunaan
ultrasonografi 3 dimensi lebih diunggulkan karena dapat mendeteksi berbagai karakteristik seperti adanya fisura palpebra yang lebih turun, mikrognatia, bentuk telinga yang lebih turun ataupun mikrotia.[12,14] Ultrasonografi juga dapat mendeteksi adanya polihidramnion, yang dapat mengarahkan kebutuhan akan pemeriksaan penunjang lainnya.[5,8] Amniosentesis dapat dilakukan untuk mengidentifikasi adanya mutasi gen dan menyingkirkan kelainan disostosis wajah lainnya seperti Sindrom Goldenhar atau Sindrom Nager.[4,8] Pada keluarga dengan risiko tinggi perlu disarankan untuk konseling genetik.[5]
Penatalaksanaan
Sebuah tim multidisiplin dilibatkan untuk mengkoordinasikan tatalaksana dalam bidang oral, okular, dental, pediatrik serta kraniofasial.[4] Secara umum, tatalaksana dibagi dalam 3 fase.[4,5] Fase pertama yaitu saat lahir sampai usia 2 tahun, penanganan jalan nafas dan masalah feeding merupakan fokus utama.[19] Fase kedua yaitu usia 2 sampai 12 tahun, mulai dilakukan terapi bicara dan rekonstruksi bagian atas wajah baik dengan transplantasi tulang maupun tandur tulang yang tervaskularisasi. Fase ketiga yaitu usia 13 sampai 18 tahun, bedah ortognatik dengan revisi lanjutannya atau transplantasi tulang kembali dilakukan.[2,5] Tatalaksana tindakan pembedahan pada pasien sindrom treacher collins dapat dilakukan sesuai usia pasien (Gambar 3).

Atrwuy JUisctNment
Slecer study
TreclMXwtcmy vs. Mundibuliir du∣nκiιon
Eyc exam, Iarsorrlupiiy (if needed)
Repeat sleep study
Repent distraction CcptuiJomctiK Cvaluitiun
Otcpbktfy - ! «t singe
Orthngnalbic surgery
l-at gr∏fι JngzniKroviwruIar free flap
Feeding, grcι* tħ evaluation
Muniiw JcnirtXMU oral hygiene
Begin rrmov∙Mc BAHA
Cleft palate repair age I
Repair of orbits, zygoma
Otoplasty - Ind stage
Eyelid repair
Pcnruneni BAHA Irnplaiilntion
Pre⅞ιι∏pαιl πrtfιodoπtics
Gambar 3. Skema Tatalaksana Komprehensif Multidisiplin Sesuai Usia Pasien[2]
Jalan Nafas dan Mandibula
Rencana preoperatif dan evaluasinya harus dimulai sedini mungkin. Umumnya saat lahir, pasien sindrom treacher collins membutuhkan trakeostomi untuk menjaga patensi nafas.[2,4] Hal ini disebabkan obstruksi pernafasan merupakan masalah utama yang sering menyebabkan apnea tidur obstruktif bahkan berujung pada insufisiensi pernafasan yang mengancam nyawa.[1] Sebuah studi oleh Plomp et al menemukan bahwa sebanyak 54% pasien mengalami apnea tidur derajat moderat.[1,2] Pada kasus yang berat, tim bedah kraniofasial segera mengevaluasi pasien yang dirawat di ruangan intensif neonatal.
Saturasi oksigen penting dinilai karena lidah pasien umumnya lebih besar dibandingkan rongga mulut disertai adanya mikrognatia, sehingga rentan terjadi desaturasi saat dalam posisi supinasi.[4] Pasien disarankan dalam posisi pronasi atau dekubitus sebagai tatalaksana awal untuk mencegah hal tersebut.[4,19] Pemeriksaan penunjang
lain seperti polisomnografi, laringoskopi atau bronkoskopi seringkali dibutuhkan.[1,2] Pemeriksaan tersebut dapat menentukan apakah penyebab sentral atau obstruktif yang mengakibatkan apnea, serta derajat dari obstruksinya.[19] Kesulitan dalam feeding juga harus dievaluasi dan disertai dengan pemberian nutrisi melalui parenteral atau enteral.[19]
Tatalaksana operatif diindikasikan jika jalan nafas terganggu akibat obstruksi lidah atau retrognatia dan hipoplasia mandibula, sehingga tindakan konservatif seperti perubahan posisi saja tidak mencukupi.[5] Prosedur operasi yang dilakukan adalah osteogenesis mandibula distraksi, operasi adhesi lidah-bibir, dan trakeostomi.[4] Tujuan utama dari pembedahan adalah dekanulasi atau pencegahan dari trakeostomi serta mengurangi derajat apnea tidur obstruktif.[4] Walaupun tindakan untuk memperbaiki maloklusi atau estetik wajah dapat dilakukan pada usia dini, namun koreksi tersebut jarang bertahan hingga dewasa, sehingga
perlu dilakukan ulang kembali secara bertahap pada usia dewasa.[5,19]
Dental dan Palatum
Celah palatum terjadi pada hampir 1/3 pasien dengan sindrom treacher collins.[16,20] Walaupun belum ada data yang merekomendasi waktu tertentu untuk operasi celah palatum pada pasien, namun perlu diketahui bahwa terdapat peningkatan resiko fistula setelah operasi akibat perfusi mukosa yang kurang optimal.[20] Selain itu pada perawatan post-operative juga perlu dilakukan rehabilitasi bicara dan bahasa secara berkala.[19,20]
Masalah lain yang sering ditemukan adalah maloklusi, dimana sebanyak 94% pasien memiliki malposisi gigi anterior.[16] Pengawasan ortodontis dimulai saat gigi permanen sudah tumbuh lengkap dan tindakan ortognatis dapat dilakukan saat remaja.[12] Evaluasi higienitas gigi dan oral penting dilakukan sejak dini karena studi menemukan bahwa pasien sindrom treacher collins memiliki kelainan pada kelenjar saliva yang rentan menyebabkan mulut kering dan meningkatkan risiko karies.[16]
Periorbital
Jaringan periorbital yang hipoplastik merupakan salah satu karakteristik patognomonis pada pasien.[2,5] Intervensi pembedahan diutamakan untuk meminimalisir resiko desikasi kornea dan jaringan parut, di samping untuk memperbaiki deformitas estetik.[11] Teknik yang digunakan untuk memperbaiki abnormalitas kelopak mata bawah antara lain adalah Z-plasti, tandur muskulokutaneus dan kantopeksi.[4] Namun, kekurangan dari hampir seluruh teknik tersebut adalah bekas jaringan parut yang tampak jelas dan deformitas permukaan kulit yang kurang baik.[4]
Tatalaksana hipoplasia zigomatiko-malar umumnya dilakukan dengan transplantasi tulang pada saat anak berusia di atas 7 tahun.[8] Namun karena tingkat resorpsi transplan tulang tinggi pada regio malar, maka beberapa studi merekomendasikan penggunaan transplan tervaskularisasi seperti tandur arteri temporal osteoperiosteal.[17] Selain itu, teknik operasi lain untuk merekonstruksi wajah bagian tengah adalah dengan teknik advancement Lefort I atau II untuk mereposisi maksila ke bentuk normal.[4,5]
Telinga dan pendengaran
Deformitas pada aurikula eksternal umumnya disertai stenosis atau atresia pada meatus auditorik eksternal.[14] Akibat dari malformasi tersebut, sebanyak 96% pasien mengalami berbagai derajat tuli.[8] Tingkat pendengaran yang berkurang juga berdampak pada kelainan bicara maupun perkembangan oral pasien.[14] Sehingga pasien harus dievaluasi secara komprehensif oleh dokter spesialis telinga hidung tenggorok, audiologis, ahli bicara serta ahli bahasa patologis.[8]
Berkaitan dengan rekonstruksi telinga, teknik autologus juga direkomendasikan sebagai metode yang umum dilakukan dengan implan polietilen.[1,4] Namun belum ada studi yang merekomendasikan usia tertentu untuk melakukan pembedahan telinga pada pasien sindrom treacher collins. Selain itu, untuk merekonstruksi garis rambut yang terletak
lebih rendah, tindakan pengangkatan rambut dengan laser dapat dilakukan.[1] Selanjutnya, rencana multidisipin untuk menentukan letak dan waktu penanaman alat bantu pendengaran harus dilakukan terutama jika operasi mikrotia akan dilakukan.[4] Hal ini disebabkan kedua prosedur tersebut terletak pada regio anatomis yang sama.[1] Penanganan kelainan pendengaran umumnya dengan pemasangan alat bantu pendengaran jenis bone-anchored sebelum operasi rekonstruksi telinga definitif dilakukan.[4,19] Hal ini bertujuan untuk mencegah keterlambatan perkembangan bahasa pasien, selama menunggu tindakan operasi rekonstruksi telinga bertahap dilakukan.[19]
SIMPULAN
Sindrom treacher collins merupakan suatu kumpulan penyakit kompleks yang berdampak pada banyak fungsi pasien. Etiologi sindrom ini disebabkan oleh kelainan pada kromosom 5q32-q33.1 sehingga muncul mutasi delesi pada gen TCOF1. Pasien yang didiagnosis harus segera dirujuk pada pusat khusus yang dapat melibatkan tim multidisipilin. Penting untuk diketahui bahwa walaupun tidak ada terapi yang dapat menyembuhkan sindrom ini, namun prognosis pasien dapat sangat berbeda sesuai derajat gejala yang dialami setiap individu. Perkembangan bahasa, pendengaran, dan pertumbuhan struktural tulang dapat sangat berpengaruh bahkan dapat menyebabkan keterlambatan tumbuh kembang jika intervensi tidak dilakukan sejak dini. Mayoritas pasien tidak mengalami gangguan saraf sehingga tatalaksana rekonstruktif penting untuk menunjang perkembangan sosial dan psikologis pasien. Teknik kraniofasial merupakan protokol
penanganan utama, walaupun jenis, waktu dan teknik operasi mandibula masih terus berkembang. Pasien dan keluarga pasien perlu diberikan edukasi akan adanya operasi bertahap pada masa kanak-kanak, bahkan dimulai sangat dini saat masih neonatus. Penilaian patensi jalan nafas merupakan prioritas utama, diikuti dengan perbaikan operatif orofaringeal serta rekonstruksi wajah bagian tengah. Sedangkan pada saat anak beranjak dewasa dilakukan perbaikan aurikula maupun pemasangan implan alat bantu pendengaran. Tahap selanjutnya diikuti oleh koreksi dental dan ortodontis pada waktu perbaikan struktur tulang wajah sudah definitif. Perkembangan studi perlu dilakukan untuk menentukan pendekatan yang lebih tepat dalam tatalaksana sindrom treacher collins.
DAFTAR PUSTAKA
-
1. Aljerian A, Gilardino MS. Treacher Collins Syndrome. Clin Plast Surg [Internet]. 2018;1–9. Available from:
https://doi.org/10.1016/j.cps.2018.11.005
-
2. Chang CC, Steinbacher DM. Treacher Collins Syndrome. Semin Plast Surg. 2012;26(2):83–90.
-
3. Vesna A. Treacher Collins Syndrome. Int Biol Biomed J. 2017;3(4):1–5.
-
4. Cobb ARM, Green B, Gill D, Ayliffe P, Lloyd TW, Bulstrode N, et al. The surgical management of Treacher Collins syndrome. Br J Oral Maxillofac Surg [Internet]. 2014;1–9. Available from: http://dx.doi.org/10.1016/j.bjoms.2014.02.007
-
5. Havlik RJ. Treacher Collins Syndrome. In: Thorne CH, editor. Grabb and Smith’s Plastic
Surgery. 7th ed. New York: Lippincott Williams & Wilkins; 2014. p. 295–311.
-
6. Zelenik K, Kominek P. Mild Form of Treacher Collins Syndrome Imitating Juvenile
Otosclerosis. Case Rep Pediatr. 2012;2012:1–3.
-
7. Dixon J, Jones N, Sandell L, Jayasinghe S, Crane J, Rey J. Tcof1_Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial
abnormalities. Proc Natl Acad Sci.
2006;103(36):13403–8.
-
8. Mehrotra D, Hasan M, Pandey R, Kumar S. Clinical spectrum of Treacher Collins syndrome. J Oral Biol Craniofacial Res [Internet]. 2011;1(1):36–40. Available from:
http://dx.doi.org/10.1016/S2212-4268(11)60009-2
-
9. Sakai D, Dixon J, Achilleos A, Dixon M, Trainor PA. Prevention of Treacher Collins syndrome craniofacial anomalies in mouse models. Nat Commun [Internet]. 2016;7:1–13. Available from: http://dx.doi.org/10.1038/ncomms10328
-
10. Lau M, Kwong E, Lai K, Li J, Ho J, Chan T, et al. Pathogenesis of POLR1C-dependent Type 3 Treacher Collins Syndrome revealed by a zebrafish model. BBA Mol Basis Dis. 2016;1862(6):1147–58.
-
11. Sharma R, Sharma B, Babber M, Singh S, Jain G. Treacher Collins syndrome: A case report and review of ophthalmic features. Taiwan J Ophthalmol [Internet]. 2016;1(4):2–5. Available from: http://dx.doi.org/10.1016/j.tjo.2016.07.002
-
12. Shetty SB, Thomas A, Pidamale R. Treacher Collins Syndrome: A Case Report and a Brief Review on Diagnostic Aids. Int J Clin Pediatr Dent. 2011;4(December):235–9.
-
13 .Jensen-Steed G. Treacher Collins Syndrome. Adv Neonatal Care. 2011;11(6):389–94.
-
14 . Kumar T, Puri G, Konidena A, Arora N. Treacher Collins syndrome: A case report and review of literature. J Indian Acad Oral Med Radiol. 2015;27(3):25–8.
-
15 .Kummer AW. Treacher Collins Syndrome. In: Cleft Palate and Craniofacial Conditions. 4th ed. Burlington: Jones & Bartlett Learning; 2014. p. 92–4.
-
16 .Bartzela TN, Carels C, Maltha JC. Update on 13 Syndromes Affecting Craniofacial and Dental Structures. Syndr Affect Orofac Struct A Rev. 2017;8(December):1038–63.
-
17 . Martelli-junior H, Coletta RD, Miranda R, Barros L De, Swerts M, Bonan P. Orofacial features of Treacher Collins syndrome. Med Oral Patol Oral Cir Bucal. 2009;14(7):344–8.
-
18 .Konstantinidou AE, Tasoulas J, Kallipolitis G, Gasparatos S, Velissariou V, Paraskevakou H. Mandibulofacial Dysostosis ( Treacher-Collins Syndrome ) in the Fetus: Novel Association with Pectus Carinatum in a Molecularly Confirmed Case and Review of the Fetal Phenotype. Birth Defects Res. 2013;780(November):774–80.
-
19 .Thompson JT, Anderson PJ, David DJ, Fracs Þ. Treacher Collins Syndrome: Protocol
Management From Birth to Maturity. J Craniofac Surg. 2009;20(6):2028–35.
-
20 .Sumbh SG, Pagare J, Sumbh B. Treacher collins syndrome - Report of a classical case. J Cleft Lip Palate Craniofacial Anomalies.
2017;4(1):69–72.
14
Discussion and feedback